Another Side of APP: Does α-Secretase Processing Drive Fragile X Syndrome?
Quick Links
As its name suggests, amyloid precursor protein (APP) gives rise to the infamous Aβ peptide that wreaks havoc in Alzheimer’s disease. Now, a study pinpoints a pathological role for non-amyloidogenic processing of APP. Researchers reported July 15 in Neuron that soluble APPα (sAPPα) runs amok in a mouse model of Fragile X Syndrome (FXS), a neurodevelopmental disorder. In young mice, the peptide ramps up the translation of other proteins, which triggers synaptic deficits and behavioral problems, according to the paper. The researchers, led by Claudia Bagni at KU Leuven in Belgium, traced the boost in sAPPα to a rise in the expression of APP and ADAM10, aka α-secretase, the protease that generates sAPPα from APP. They also found evidence that this pathway goes awry in human cases of FXS, and proposed that blocking ADAM10 in early life may help treat the disorder.
“This [study] is significant in that it raises the intriguing possibility that abnormal APP production and processing could underlie both Fragile X, a neurodevelopmental disorder, and Alzheimer’s disease, the most common neurodegenerative disease in old age,” commented Hui Zheng of Baylor College of Medicine in Houston. She added that a bevy of questions remain unanswered.
FXS is caused by the loss of or mutations in the gene encoding Fragile X mental retardation protein (FMRP), an RNA-binding protein that turns down the translation of many genes involved in neural function. About a third of people with FXS—who have behavioral, learning, and anxiety problems—also meet the criteria for Autism Spectrum Disorder (ASD) (see Lozano et al., 2014). APP is among the 40 or so genes confirmed to be regulated by FMRP (see Westmark and Malter, 2007; Pascuito and Bagni, 2014).
Soluble APPα promotes synaptogenesis and boosts synaptic density (see Bell et al., 2008; Tyan et al., 2012; Hick et al., 2015). Both APP and sAPPα are highly expressed during periods of intense synaptogenesis in humans and in mice (see Moya et al., 1994). People with FXS reportedly have an overabundance of immature dendritic spines, which are long, skinny, and twisted (see Irwin et al., 2011).
First author Emanuela Pascuito and colleagues wondered whether too much of a good thing, sAPPα, could explain the synaptic mayhem in FXS. The researchers tracked levels of APP, sAPPα, and ADAM10 throughout postnatal development in FMR1 knockout mice, which display many of the characteristics of the syndrome. APP levels were normal until the animals reached 3 weeks of age, at which point they shot up compared to controls and remained high throughout adulthood. While the total amount of APP rose in the KO neurons, cell surface levels of APP were lower than those in normal cells, suggesting internalization or an increased shedding of APP fragments. ADAM10 and sAPPα also increased in the knockout mice at 3 weeks, though their levels returned to normal by 3 months of age.
The researchers next zoomed in on dendritic spines. They compared the shape and size of the spines on neurons isolated from normal or FMR1 KO mice, and found that spines on knockout neurons tended to be more abundant and immature-looking; they were long and thin, rather than mushroom-shaped or stubby. Knocking down APP expression in these neurons reduced the density of these wispy spines, but they flourished again when the authors added sAPPα. This suggested that the elevated levels of sAPPα in FMR1-negative mice were responsible for the shift toward immature spines.

Spindly Spines.
In FMR1 KO neurons, sAPPα compromises the structure of dendritic spines (see text for details). [Courtesy of Pascuito et al., Neuron 2015.]
Because FMR1 KO mice are known to have elevated protein synthesis, the researchers next wanted to test whether elevated sAPPα helped promote this effect. They labeled newly synthesized proteins in cortical neurons from KO versus normal mice and found that, as expected, the former pumped out more protein. However, when the researchers crossed the knockout mice with animals expressing only a single copy of APP or ADAM10, cortical neurons from the offspring made normal amounts of protein. On the other hand, adding sAPPα to neurons from any of the mouse strains boosted protein synthesis. Together, these results suggested that sAPPα helped drive exaggerated protein synthesis in the Fragile X mice.
How would elevated sAPPα raise global protein synthesis? The mGluR5 pathway is a possibility. Previous reports indicate that this metabotropic glutamate receptor and downstream MAP kinase signaling are affected in FXS, and that enhanced signaling through this receptor elevates protein synthesis. The researchers found that sAPPα enhanced MAP kinase phosphorylation of ERK1/2 in cortical neurons, and that this increase was abolished when the cells were treated with a GluR1/5 inhibitor. Bagni said that her lab is currently investigating how sAPPα switches on the GluR5 pathway.
Would inhibiting ADAM10, and thus production of sAPPα, ameliorate FXS symptoms in the mice? To find out, the researchers injected the animals with Tat-Pro709-729 ADAM10, a peptide fragment that contains part of the ADAM10 intracellular domain and interferes with its protease activity. Tat-Pro lowered sAPPα levels to normal in FMR1 KO mice. The inhibitor also normalized long-term depression (LTD), which is enhanced in FXS. Tat-Pro also reversed several learning and behavioral problems in the knockouts, including working memory deficits, hyperactivity, and poor nest building.
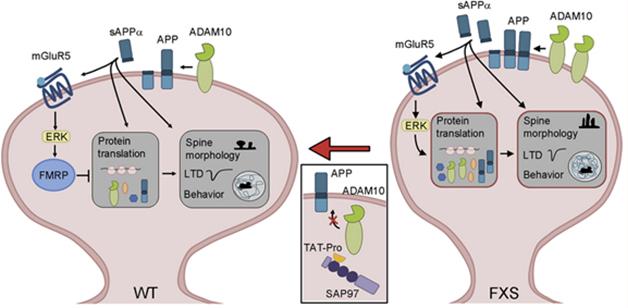
sAPPαOverdrive. In wild-type neurons (left), FMRP keeps protein translation in check. In FXS neurons, loss of FMRP takes the brakes off translation, while overproduction of sAPPα keeps the translational machinery humming. [Courtesy of Pascuito et al., Neuron 2015.]
“This is a great paper,” commented Bernadette Allinquant at INSERM in Paris. Allinquant has worked to characterize the functions of sAPPα for decades. “I’ve always thought of sAPPα as a ‘good molecule.’ It has neurotrophic and neuroprotective properties and increases LTP,” she said. “For the first time, this paper reveals that too much sAPPα at the wrong time is a bad thing.”
The researchers searched for clues that this pathway played a role in FXS in humans. They found elevated APP (but not ADAM10) in brain and lymphoblastoid cells from older patients, and elevated levels of both APP and ADAM10 in fibroblasts from younger patients. These changes paralleled the developmental regulation of these proteins in FMR1 KO mice—with APP levels high throughout adulthood, and ADAM10 only going up during younger life.
The researchers are collecting fibroblast samples, and plan to measure fluctuations in sAPPα and ADAM10 from early childhood into adulthood. Bagni wants to home in on the window during which sAPPα rises in people with FXS, and test whether giving an ADAM10 inhibitor during that time could ameliorate some symptoms of the disease. “After that critical period, ADAM10 is no longer dysregulated, and this treatment would not work,” she said. She also plansto test whether treatment of juvenile mice with the inhibitor fends off the manifestation of disease later on.
Recent studies reported elevated serum sAPPα in autism spectrum disorder and FXS patients, and Bagni envisions using such a biomarker to select and monitor patients in clinical trials (see Ray et al., 2011; Erickson et al., 2014). The elevated sAPPα in autism patients who do not have FXS raises the interesting possibility that the fragment plays a role in a host of disorders.
“These studies contribute to a growing body of knowledge suggesting that APP processing is developmentally regulated and aberrant in both fragile X and autism,” wrote Cara Westmark of the University of Wisconsin in Madison. She added that much is left to learn about the changing balance between non-amyloidogenic and amyloidogenic APP processing throughout early development and adulthood. She also cautioned that toying with ADAM10 activity or levels of APP metabolites could promote seizures, as has been demonstrated in ADAM10 knockout mice. Bagni said that the ADAM10 inhibitors merely return the protease activity to normal.—Jessica Shugart
References
Paper Citations
- Lozano R, Rosero CA, Hagerman RJ. Fragile X spectrum disorders. Intractable Rare Dis Res. 2014 Nov;3(4):134-46. PubMed.
- Westmark CJ, Malter JS. FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 2007 Mar;5(3):e52. PubMed.
- Pasciuto E, Bagni C. SnapShot: FMRP mRNA targets and diseases. Cell. 2014 Sep 11;158(6):1446-1446.e1. PubMed.
- Bell KF, Zheng L, Fahrenholz F, Cuello AC. ADAM-10 over-expression increases cortical synaptogenesis. Neurobiol Aging. 2008 Apr;29(4):554-65. Epub 2006 Dec 21 PubMed.
- Tyan SH, Shih AY, Walsh JJ, Maruyama H, Sarsoza F, Ku L, Eggert S, Hof PR, Koo EH, Dickstein DL. Amyloid precursor protein (APP) regulates synaptic structure and function. Mol Cell Neurosci. 2012 Aug;51(1-2):43-52. PubMed.
- Hick M, Herrmann U, Weyer SW, Mallm JP, Tschäpe JA, Borgers M, Mercken M, Roth FC, Draguhn A, Slomianka L, Wolfer DP, Korte M, Müller UC. Acute function of secreted amyloid precursor protein fragment APPsα in synaptic plasticity. Acta Neuropathol. 2015 Jan;129(1):21-37. Epub 2014 Nov 29 PubMed.
- Moya KL, Benowitz LI, Schneider GE, Allinquant B. The amyloid precursor protein is developmentally regulated and correlated with synaptogenesis. Dev Biol. 1994 Feb;161(2):597-603. PubMed.
- Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, Swain RA, Weiler IJ, Greenough WT. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet. 2001 Jan 15;98(2):161-7. PubMed.
- Ray B, Long JM, Sokol DK, Lahiri DK. Increased secreted amyloid precursor protein-α (sAPPα) in severe autism: proposal of a specific, anabolic pathway and putative biomarker. PLoS One. 2011;6(6):e20405. Epub 2011 Jun 22 PubMed.
- Erickson CA, Ray B, Maloney B, Wink LK, Bowers K, Schaefer TL, McDougle CJ, Sokol DK, Lahiri DK. Impact of acamprosate on plasma amyloid-β precursor protein in youth: a pilot analysis in fragile X syndrome-associated and idiopathic autism spectrum disorder suggests a pharmacodynamic protein marker. J Psychiatr Res. 2014 Dec;59:220-8. Epub 2014 Aug 19 PubMed.
Further Reading
News
Primary Papers
- Pasciuto E, Ahmed T, Wahle T, Gardoni F, D'Andrea L, Pacini L, Jacquemont S, Tassone F, Balschun D, Dotti CG, Callaerts-Vegh Z, D'Hooge R, Müller UC, Di Luca M, De Strooper B, Bagni C. Dysregulated ADAM10-Mediated Processing of APP during a Critical Time Window Leads to Synaptic Deficits in Fragile X Syndrome. Neuron. 2015 Jul 15;87(2):382-98. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Baylor College of Medicine
A role for FMRP as a translational repressor of APP has been reported earlier. Here the authors present the novel finding that ADAM10 is also a target of FMRP, and that ADAM10 levels increase when FMRP is absent. The combination of increased APP and ADAM10 synthesis in Fmr1 knockout mice leads to elevated sAPPα, which is responsible for the molecular, morphological, and functional deficits observed in the Fmr1 mutant mice and, by extension, Fragile X patients. This is significant in that it raises the intriguing possibility that abnormal APP production and APP processing could underlie both Fragile X, a neurodevelopmental disorder, and Alzheimer’s disease, the most common neurodegenerative disease in old age. However, several questions remain to be addressed: First, why does the regulation of APP and ADAM10 by FMRP only occur during a specific postnatal development stage? Second, what are the mechanisms linking sAPPα to mGluR5 signaling and protein translation? Lastly, do such sAPPα-mediated pathways also regulate neuronal function in adult and aged brains?
While not related to the current study, it is interesting to note that APP is also overproduced in another intellectual disability disorder, Down’s syndrome (DS). Therefore, it is tempting to ask whether increased APP (and sAPPα) in DS could, through similar pathways revealed here, contribute to early neurological impairment in addition to the development of AD in later ages.
University of Wisconsin
This Neuron paper by Pasciuto and colleagues describes the effects of dysregulated ADAM10-mediated processing of APP on Fragile X syndrome. Their evidence for the role of soluble APP alpha (sAPPα) in mediating immature spine, mRNA translation, and mGluR-LTD phenotypes in Fmr1KO mice dovetails nicely with recent studies regarding the role of non-amyloidogenic (α-secretase) processing of APP in autism (Sokol et al., 2006; Bailey et al., 2013; Ray et al., 2011; Lahiri et al., 2013). Their data also suggest that pharmacological inhibition of ADAM10 may be a viable therapeutic option for Fragile X.
A caveat to these findings is that APP is also regulated by amyloidogenic (β-secretase) processing. Our laboratory observed elevated levels of Aβ in older Fmr1KO mice and rescue of numerous disease phenotypes in Fmr1KO/APPHET mice, which support testing β-secretase inhibitors in Fragile X (Westmark et al., 2007). The presence of elevated Aβ levels in older mice was reproduced in the current study, which went on to show that juvenile mice exhibit reduced levels of Aβ. Abnormal Aβ accumulation has also been observed in idiopathic and Dup15 autism brains in both adults and children (Wegiel et al., 2012; Al-Ayadhi et al., 2012). These studies contribute to a growing body of knowledge suggesting that APP processing is developmentally regulated and aberrant in both Fragile X and autism. Much remains to be learned regarding: (1) APP metabolite profiles as a function of age, (2) potential alterations in proteolytic processing of APP with development, and (3) associations between APP metabolite levels and Fragile X and autism traits. The current study contributes a substantial amount of data regarding the role of sAPPα in Fragile X, particularly during the early postnatal stage of development.
From a therapeutic standpoint, it may be necessary to simultaneously modulate both α- and β-secretase processing to attain homeostatic levels of APP metabolites and rescue Fragile X phenotypes. We have observed that both overexpression and knockout of APP increases seizure propensity in juvenile Fmr1KO mice. Born and colleagues demonstrated that juvenile overexpression of APP contributes to sharp wave EEG discharges in APP transgenic mice; they did not differentiate between full-length APP and sAPPα (Born et al., 2014). And Prox and colleagues found that seizures are increased in the ADAM10 conditional knockout mouse (Prox et al., 2013). Seizures were not studied in the Pasciuto publication; thus, the role of ADAM10 and sAPPα on seizure propensity in the Fmr1KO remains to be determined. It is likely that various APP metabolites contribute to different fragile phenotypes. Of interest, numerous drugs currently under study for FXS may be effective due to off-site activities that modulate APP, Aβ, β-secretase and/or ADAM10. (Westmark et al., 2013; Erickson, 2014). For example, the acetylcholinesterase inhibitor donepezil (Aricept), which is used to treat Alzheimer’s disease, has been shown to improve cognitive-behavioral function in Fragile X (Kesler et al., 2009). Donepezil decreases Aβ levels (Dong et al., 2009) and increases ADAM10 activity and the release of sAPPα (Zimmerman, 2005).
Overall, this exciting publication by the Bagni laboratory demonstrates important roles for ADAM10 and sAPPα in Fragile X pathology and provides a strong impetus for future studies to define the differential roles of APP metabolites during development and the therapeutic efficacy of secretase drugs in Fragile X syndrome.
References:
Sokol DK, Chen D, Farlow MR, Dunn DW, Maloney B, Zimmer JA, Lahiri DK. High levels of Alzheimer beta-amyloid precursor protein (APP) in children with severely autistic behavior and aggression. J Child Neurol. 2006 Jun;21(6):444-9. PubMed.
Bailey AR, Hou H, Song M, Obregon DF, Portis S, Barger S, Shytle D, Stock S, Mori T, Sanberg PG, Murphy T, Tan J. GFAP expression and social deficits in transgenic mice overexpressing human sAPPα. Glia. 2013 Sep;61(9):1556-69. PubMed.
Ray B, Long JM, Sokol DK, Lahiri DK. Increased secreted amyloid precursor protein-α (sAPPα) in severe autism: proposal of a specific, anabolic pathway and putative biomarker. PLoS One. 2011;6(6):e20405. Epub 2011 Jun 22 PubMed.
Lahiri DK, Sokol DK, Erickson C, Ray B, Ho CY, Maloney B. Autism as early neurodevelopmental disorder: evidence for an sAPPα-mediated anabolic pathway. Front Cell Neurosci. 2013;7:94. PubMed.
Westmark CJ, Malter JS. FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 2007 Mar;5(3):e52. PubMed.
Westmark CJ, Westmark PR, O'Riordan KJ, Ray BC, Hervey CM, Salamat MS, Abozeid SH, Stein KM, Stodola LA, Tranfaglia M, Burger C, Berry-Kravis EM, Malter JS. Reversal of fragile X phenotypes by manipulation of AβPP/Aβ levels in Fmr1KO mice. PLoS One. 2011;6(10):e26549. PubMed.
Wegiel J, Frackowiak J, Mazur-Kolecka B, Schanen NC, Cook EH, Sigman M, Brown WT, Kuchna I, Nowicki K, Imaki H, Ma SY, Chauhan A, Chauhan V, Miller DL, Mehta PD, Flory M, Cohen IL, London E, Reisberg B, de Leon MJ, Wisniewski T. Abnormal intracellular accumulation and extracellular Aβ deposition in idiopathic and Dup15q11.2-q13 autism spectrum disorders. PLoS One. 2012;7(5):e35414. PubMed.
Al-Ayadhi LY, Ben Bacha AG, Kotb M, El-Ansary AK. A novel study on amyloid β peptide 40, 42 and 40/42 ratio in Saudi autistics. Behav Brain Funct. 2012 Jan 13;8:4. PubMed.
Born HA, Kim JY, Savjani RR, Das P, Dabaghian YA, Guo Q, Yoo JW, Schuler DR, Cirrito JR, Zheng H, Golde TE, Noebels JL, Jankowsky JL. Genetic suppression of transgenic APP rescues Hypersynchronous network activity in a mouse model of Alzeimer's disease. J Neurosci. 2014 Mar 12;34(11):3826-40. PubMed.
Prox J, Bernreuther C, Altmeppen H, Grendel J, Glatzel M, D'Hooge R, Stroobants S, Ahmed T, Balschun D, Willem M, Lammich S, Isbrandt D, Schweizer M, Horré K, De Strooper B, Saftig P. Postnatal Disruption of the Disintegrin/Metalloproteinase ADAM10 in Brain Causes Epileptic Seizures, Learning Deficits, Altered Spine Morphology, and Defective Synaptic Functions. J Neurosci. 2013 Aug 7;33(32):12915-28, 12928a. PubMed.
Westmark CJ, Berry-Kravis EM, Ikonomidou C, Yin JC, Puglielli L. Developing BACE-1 inhibitors for FXS. Front Cell Neurosci. 2013;7:77. Epub 2013 May 28 PubMed.
Erickson CA, Ray B, Maloney B, Wink LK, Bowers K, Schaefer TL, McDougle CJ, Sokol DK, Lahiri DK. Impact of acamprosate on plasma amyloid-β precursor protein in youth: a pilot analysis in fragile X syndrome-associated and idiopathic autism spectrum disorder suggests a pharmacodynamic protein marker. J Psychiatr Res. 2014 Dec;59:220-8. Epub 2014 Aug 19 PubMed.
Kesler SR, Lightbody AA, Reiss AL. Cholinergic dysfunction in fragile X syndrome and potential intervention: a preliminary 1H MRS study. Am J Med Genet A. 2009 Mar;149A(3):403-7. PubMed.
Dong H, Yuede CM, Coughlan CA, Murphy KM, Csernansky JG. Effects of donepezil on amyloid-beta and synapse density in the Tg2576 mouse model of Alzheimer's disease. Brain Res. 2009 Dec 15;1303:169-78. Epub 2009 Sep 30 PubMed.
Zimmermann M, Borroni B, Cattabeni F, Padovani A, Di Luca M. Cholinesterase inhibitors influence APP metabolism in Alzheimer disease patients. Neurobiol Dis. 2005 Jun-Jul;19(1-2):237-42. PubMed.
Make a Comment
To make a comment you must login or register.