ALS-FTD Mouse Model Develops Motor Neuron Disease
Quick Links
In the November 9 Proceedings of the National Academy of Sciences, scientists led by Mervyn Monteiro, University of Maryland School of Medicine, Baltimore, describe two new models of amyotrophic lateral sclerosis/frontotemporal dementia. The researchers expressed mutant versions of the ubiquilin 2 gene in neurons of mice. The animals developed learning deficits, as did other UBQLN2 models, but in addition, their motor neurons degenerated and aggregates of TDP-43 accumulated in their spinal cords. These animals, which recapitulate the TDP-43 pathology that underlies ALS/FTD better than any other model to date, may help scientists understand the links between UBQLN2 and proteostasis, as well as between TDP-43 pathology and motor neuron disease, said Monteiro. Other researchers cautioned that the promoter used to overexpress UBQLN2 may underlie the neurodegeneration, not UBQLN2 itself.
“This is a major improvement to existing rodent models of ALS-FTD,” wrote Mark Verheijen, University Medical Center Utrecht, The Netherlands, to Alzforum. “The finding directly relates a major ALS disease pathway—impaired proteostasis—to a key neuropathological feature in ALS,” namely TDP-43 pathology, he wrote. Verheijen was not involved in the work.
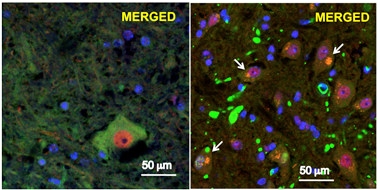
Renegade Proteins.
In motor neurons from a mouse model of ALS/FTD (right), P497S UBQLN2 (green) aggregates, TDP-43 (red) vacates the nucleus, and the two co-localize (white arrows). Left shows normal neurons. [Image courtesy of Le et al., 2016. PNAS.]
Ubiquilin 2 binds proteins tagged with ubiquitin and shuttles them to the proteasome for disposal (for a review, see Lee and Brown, 2012). This process is crucial for clearing protein aggregates (Brown and Kaganovitch, 2016). Ubiquilin 2 also helps deliver autophagosomes to lysosomes, which degrade unwanted proteins (N’Diaye et al., 2009; Rothenberg et al., 2010). In 2011, researchers linked variants in the UBQLN2 gene with familial ALS/FTD (Deng et al., 2011). Since then, UBQLN2 mutations have been identified in sporadic ALS and FTD, accounting for an estimated 1 to 2 percent of cases, respectively (Synofzik et al., 2012). Patients who carry a single copy of the mutated gene accumulate ubiquilin 2-positive inclusions in neurons of the hippocampus, and TDP-43 aggregates in motor neurons in the spinal cord. The disease variants are mostly missense mutations that concentrate around the region coding for the protein’s central domain (Williams et al., 2012).
To study how UBQLN2 mutations lead to ALS and FTD, researchers have generated several mouse and rat models that overexpress the mutated human gene. Some of the animals develop cognitive deficits, ubiquilin inclusions, and neuron loss in the brain (Wu et al., 2015; Gorrie et al., 2014), but so far, none have motor neuron disease. One strain performs poorly on the rotarod test and clasps its paws more than normal animals, both signs of potential motor neuron problems, but the physiological reason for these behaviors is unclear (Ceballos-Diaz et al., 2015). Monteiro and colleagues aimed to create a UBQLN2 mouse model that more fully mirrored human disease.
First authors Nhat Le, Lydia Chang, and Irina Kovlyagina used a neuron-specific Thy1.2 expression cassette to produce the P497S, P506T, or wild-type variants of UBQLN2 in the mouse brain and spinal cord. Mice expressed their own form of the protein, plus about 70 to 80 percent more of the human versions. Both the P497S and P506T variants have been linked to ALS/FTD.
Le and colleagues weighed the mice and measured their motor functions biweekly from two to eight months of age. Those that expressed the P497S or P506T variants gained weight normally until 18 and 26 weeks, respectively, then fell behind. At the end of the study, they were about 30 percent lighter than control mice expressing the normal transgene. At six weeks, the P506T mice began to lose grip strength and could not hang onto a rotating rod as long as did controls. For P497S mice this started at 12 weeks. Both strains also clasped their paws more, and in some animals the hind limbs became paralyzed.
At two and four months, the researchers tested learning and memory in the animals using a novel-object-recognition task and a Y-maze. Like controls, two- and four-month-old mutant UBQLN2 carriers preferred to venture down the novel arm of the Y-maze, suggesting normal learning and memory. However, at four months, the mice had trouble distinguishing between novel and familiar objects placed in the testing arena, suggesting their memory was impaired. It is unclear if these problems worsen as the mice age since older animals were too weak to test.
Mice overexpressing the mutant UBQLN2 died young, at an average 246 days for the P506T line and 305 for the P497S mice. Animals that expressed the wild-type transgene outlived the 500-day study without motor difficulties.
In postmortem analysis, the researchers found an age-dependent buildup of UBQLN inclusions in neurons of the brain. These inclusions populated layers V and VI of the cortex, parts of the dentate gyrus, CA1 region of the hippocampus, brainstem, and striatum. The pattern resembled that seen in other UBQLN2 rodent models and in ALS/FTD patients with UBQLN2 mutations, the authors wrote. Inclusions tested positive for ubiquitin and Thioflavin S.
The authors then tested for TDP-43 in the motor neurons of the spinal cord. Similar to what is seen in humans, the protein vacated the nucleus to form granular inclusions in the cytoplasm, which also contained ubiquilin 2 (see image above). Mice that expressed no transgene, or the wild-type version of human UBQLN2, had no TDP-43 pathology and few ubiquilin 2 inclusions, which according to Monteiro can form as normal animals age. Researchers detect TDP-43 pathology in 97 percent of human ALS cases but not in the SOD1 mouse models that are commonly used in ALS research. Some models have TDP-43 inclusions but retain most of the protein in the nucleus, Monteiro added. “Because nuclear clearance could be mechanistically linked to ALS pathogenesis, recapitulating this phenomenon in models is important,” he suggested.
The researchers then compared postmortem tissue samples of three- and eight-month-old animals. Older P506T and P497S mice had fewer motor neurons in the ventral horn of the spinal cord and in the CA1 and dentate gyrus of the hippocampus. They sported smaller calf muscles, thinner muscle fibers, and fewer large-caliber axons, suggesting muscle wasting and degeneration. Synaptophysin and α-bungarotoxin co-stained less often at the neuromuscular junctions, suggesting reduced innervation. Taken together, the results point to motor neuron disease in these animals.
Monteiro is unsure why motor neurons degenerate in these but not other UBQLN2 models. He suggested that the levels of protein expression in other models were lower because they used the CamK2α or the endogenous murine UBQLN2 promoter. Alternatively, it could be because the regional expression of the transgene was more restricted to the brain, he said. Along those lines, Manuela Neumann, University of Tübingen, Germany, cautioned that the Thy1.2 promoter leads to abnormally high expression of transgenes in the spinal cord, which could lead to motor difficulties independent of the mutations themselves. “As always, one has to be very cautious with interpreting results from overexpression models, particularly those with ectopic expression,” she said. Teepu Siddique, Northwestern University, Chicago, echoed the concern, noting that motor paralysis or weakness can be induced by expressing genes unrelated to ALS using the Thy1.2 promoter (Götz et al., 2000). Monteiro pointed out that control mice expressed wild-type UBQLN2 driven by the Thy1.2 promoter, yet did not develop motor neuron disease.
Neumann found it interesting that UBQLN2 overexpression appeared to kill cells even in regions that lacked TDP-43 pathology, such as the hippocampus. This finding jibes with other UBQLN2 models where cell death occurs in the absence of TDP-43 pathology. “This suggests that TDP-43 aggregation per se might not be a crucial step mediating cell death in ALS/FTD cases with UBQLN2 mutations,” said Neumann.
Other scientists thought the mice could be illuminating. “These UBQLN2 mutant mice can help researchers investigate the exact time course of, and connection between, histopathological features [of ALS/FTD], such as UBQLN2 inclusion formation, TDP43 pathology, muscle denervation, neuronal cell loss, and gliosis,” wrote Gregor Bieri, Stanford University School of Medicine in California. “Further, they could be used specifically to test potential therapeutic interventions that target UBQLN2 expression or accumulation,” he said.
Verheijen would like to know if RNA processing and transport is changed in these mice. After all, both have been linked to ALS/FTD through RNA-binding proteins such as TDP43 and FUS, and toxic RNA species generated from expansions in the C9ORF72 gene. Monteiro plans to make the mice available, possibly through the Jackson Laboratory in Bar Harbor, Maine.—Gwyneth Dickey Zakaib
References
Paper Citations
- Lee DY, Brown EJ. Ubiquilins in the crosstalk among proteolytic pathways. Biol Chem. 2012 May;393(6):441-7. PubMed.
- Brown R, Kaganovich D. Look Out Autophagy, Ubiquilin UPS Its Game. Cell. 2016 Aug 11;166(4):797-9. PubMed.
- N'Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, Brown EJ. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009 Feb;10(2):173-9. Epub 2009 Jan 16 PubMed.
- Rothenberg C, Srinivasan D, Mah L, Kaushik S, Peterhoff CM, Ugolino J, Fang S, Cuervo AM, Nixon RA, Monteiro MJ. Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum Mol Genet. 2010 Aug 15;19(16):3219-32. PubMed.
- Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, Jiang H, Hirano M, Rampersaud E, Jansen GH, Donkervoort S, Bigio EH, Brooks BR, Ajroud K, Sufit RL, Haines JL, Mugnaini E, Pericak-Vance MA, Siddique T. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011 Sep 8;477(7363):211-5. PubMed.
- Synofzik M, Maetzler W, Grehl T, Prudlo J, Vom Hagen JM, Haack T, Rebassoo P, Munz M, Schöls L, Biskup S. Screening in ALS and FTD patients reveals 3 novel UBQLN2 mutations outside the PXX domain and a pure FTD phenotype. Neurobiol Aging. 2012 Dec;33(12):2949.e13-7. PubMed.
- Williams KL, Warraich ST, Yang S, Solski JA, Fernando R, Rouleau GA, Nicholson GA, Blair IP. UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol Aging. 2012 Oct;33(10):2527.e3-2527.e10. PubMed.
- Wu Q, Liu M, Huang C, Liu X, Huang B, Li N, Zhou H, Xia XG. Pathogenic Ubqln2 gains toxic properties to induce neuron death. Acta Neuropathol. 2015 Mar;129(3):417-28. Epub 2014 Nov 12 PubMed.
- Gorrie GH, Fecto F, Radzicki D, Weiss C, Shi Y, Dong H, Zhai H, Fu R, Liu E, Li S, Arrat H, Bigio EH, Disterhoft JF, Martina M, Mugnaini E, Siddique T, Deng HX. Dendritic spinopathy in transgenic mice expressing ALS/dementia-linked mutant UBQLN2. Proc Natl Acad Sci U S A. 2014 Oct 7;111(40):14524-9. Epub 2014 Sep 22 PubMed.
- Ceballos-Diaz C, Rosario AM, Park HJ, Chakrabarty P, Sacino A, Cruz PE, Siemienski Z, Lara N, Moran C, Ravelo N, Golde TE, McFarland NR. Viral expression of ALS-linked ubiquilin-2 mutants causes inclusion pathology and behavioral deficits in mice. Mol Neurodegener. 2015 Jul 8;10:25. PubMed.
- Götz J, Barmettler R, Ferrari A, Goedert M, Probst A, Nitsch RM. In vivo analysis of wild-type and FTDP-17 tau transgenic mice. Ann N Y Acad Sci. 2000;920:126-33. PubMed.
Further Reading
Primary Papers
- Le NT, Chang L, Kovlyagina I, Georgiou P, Safren N, Braunstein KE, Kvarta MD, Van Dyke AM, LeGates TA, Philips T, Morrison BM, Thompson SM, Puche AC, Gould TD, Rothstein JD, Wong PC, Monteiro MJ. Motor neuron disease, TDP-43 pathology, and memory deficits in mice expressing ALS-FTD-linked UBQLN2 mutations. Proc Natl Acad Sci U S A. 2016 Nov 22;113(47):E7580-E7589. Epub 2016 Nov 9 PubMed. Correction.
Annotate
To make an annotation you must Login or Register.
Comments
UMC Utrecht
There have been lots of studies that try to show how TDP-43 inclusions arise, what their role in disease is, and how (aggregated) TDP-43 can be effectively cleared from cells. There is still a lot of debate on what the exact contribution of different protein degradation pathways is. A link between disturbed proteostasis and TDP-43 pathology has always been clear, but to my knowledge, previously generated UBQLN2 mutant mice did not develop TDP-43 inclusions at all (and of course not all available rodent models of ALS do). Therefore, it would be very interesting to find out how UBQLN2 controls this process in vivo and to study the consequences of this secondary TDP-43 aggregation.
University of Tübingen and DZNE AG Neumann
These are certainly interesting novel mouse models for future studies on ALS/FTD pathogenesis. Several aspects seen in these novel mouse models confirm previous findings in comparable transgenic mouse and rat models, namely that overexpression of UBQLN2 leads to accumulation of ubiquilin 2 together with other proteins of the ubiquitin/proteasome pathway consistently in the hippocampus, which results in cell loss and impaired cognitive function (Gorrie et al., 2014; Wu et al., 2015; Huang et al., 2016). Notably, these novel mouse lines also show an additional motor neuron phenotype and motor neuron degeneration, which is very likely simply due to the fact that the use of Thy1 as a promoter in these novel mice leads to a very strong expression of the transgene in the spinal cord (other transgenic models which either use CamKII or the endogenous UBQLN2 promoter, as also discussed by the authors). As always, one has to be very cautious with interpreting results from overexpressing models, and particularly those with ectopic expression, with respect to their relevance in understanding the human diseases.
Nevertheless, and independent from this concern, it is very interesting that in the spinal cord, the overexpression of UBQLN2 mutations leads to TDP-43 pathology. This is not the case in other regions, such as the hippocampus, which is also affected by cell loss in these mice in agreement with lack of TDP-43 pathology in other UBQLN2 transgenic models. This suggests that TDP-43 aggregation per se might not be a crucial step required for mediating cell death in ALS/FTD cases with UBQLN2 mutations—at least not in all cell types.
It will be interesting to see follow-up investigations in these mouse lines to further define the TDP-43 pathology (e.g., are the cytoplasmic inclusions composed of hyperphosphorylated TDP-43?) and particularly the link between TDP-43 accumulation and cell death of motor neurons (e.g., is it a bystander or directly linked to cell death?) and to dissect the “TDP-43 independent” mechanisms of toxicity upon overexpression of UBQLN2 mutations.
References:
Gorrie GH, Fecto F, Radzicki D, Weiss C, Shi Y, Dong H, Zhai H, Fu R, Liu E, Li S, Arrat H, Bigio EH, Disterhoft JF, Martina M, Mugnaini E, Siddique T, Deng HX. Dendritic spinopathy in transgenic mice expressing ALS/dementia-linked mutant UBQLN2. Proc Natl Acad Sci U S A. 2014 Oct 7;111(40):14524-9. Epub 2014 Sep 22 PubMed.
Wu Q, Liu M, Huang C, Liu X, Huang B, Li N, Zhou H, Xia XG. Pathogenic Ubqln2 gains toxic properties to induce neuron death. Acta Neuropathol. 2015 Mar;129(3):417-28. Epub 2014 Nov 12 PubMed.
Huang B, Wu Q, Zhou H, Huang C, Xia XG. Increased Ubqln2 expression causes neuron death in transgenic rats. J Neurochem. 2016 Oct;139(2):285-293. PubMed.
Stanford University - School of Medicine
Li et al. have developed a series of new mouse lines in which either wild-type (WT) or two ALS/FTD-associated mutated forms (P497S and P506T) of the human UBQLN2 gene are overexpressed broadly in neurons. The expression of the mutated but not the WT protein leads to the formation and progressive accumulation of UBQLN2-positive inclusions in several brain and spinal cord regions. This model recapitulates several key degenerative phenotypes of human ALS, such as a loss of lower motor neurons, muscle denervation/atrophy, gliosis, and impairments in muscle strength and motor tasks. Most excitingly, the model develops TDP43 pathology (nuclear clearance and formation of cytoplasmic inclusions) in motor neurons, a key histopathological hallmark observed in a majority of human ALS cases. Additionally, the authors report neuronal cell loss in other CNS areas such as the hippocampus and associated impairments in learning and memory tasks. Interestingly, no TDP43 pathology was observed in neurons other than the spinal cord motor neurons.
These new mouse lines could prove to be a valuable in vivo tool to decipher how UBQLN2 mutations cause the degenerative disease phenotypes. The UBQLN2 mutant mice can be used to investigate the exact time course of, and connection between, histopathological features such as UBQLN2 inclusion formation, TDP43 pathology, muscle denervation, neuronal cell loss, and gliosis. Further, they could be used to test potential therapeutic interventions targeting UBQLN2 expression or accumulation specifically.
More broadly, a model that faithfully reproduces key hallmarks of ALS/FTD potentially could be used to test more global therapeutic interventions, such as targeting mislocalized TDP43 in the absence of TDP43 overexpression, or therapeutics aimed at promoting neuronal survival or preventing neuroinflammation.
Make a Comment
To make a comment you must login or register.