In ALS, Astrocyte TGF-β1 Pulls Rug From Under Motor Neurons
Quick Links
Researchers have long wondered why glia and other immune cells turn their backs on motor neurons as amyotrophic lateral sclerosis progresses. Now, they may be closer to figuring it out. Astrocytes produce transforming growth factor-β1, which deactivates microglia just when those neurons need them most, according to a paper online in Cell Reports April 16. The finding partially solves a longstanding puzzle in the field: Astrocytes were known to secrete something that caused the death of motor neurons, and TGF-β1 looks to be a component of that killer signal, said senior author Koji Yamanaka of Nagoya University in Japan.

Powering down:
The microglia of mSOD1 mice (left) are normally active, making CD68 (yellow), but stay quiescent in mSOD1 mice that produce excess TGF-β1 (right). [Image courtesy of Cell Reports, Endo et al.]
Yamanaka had previously found that astrocytes played a role in disease progression in mice carrying the mutant ALS gene SOD1. Selectively deleting mutant SOD1 in astrocytes slowed disease in those animals (Yamanaka et al., 2008). In the current work, first author Fumito Endo and colleagues focused on the anti-inflammatory factor TGF-β1 because astrocytes produce it in response to injury, and it ticks up in the blood and cerebrospinal fluid of people with ALS (Houi et al., 2002; Itzecka et al., 2002).
When Endo examined spinal-cord tissue from mSOD1 mice and from people who died of ALS, he confirmed that both contained more TGF-β1 than control tissue. In particular, the astrocytes were awash in TGF-β1. To examine how TGF-β1 affected the disease, Endo crossed the mSOD1 mice with a line engineered to make more of the growth factor in astrocytes. The authors predicted the growth factor might help the mice, because other studies had found that bumping up TGF-β1 signaling was neuroprotective (see Dec 2003 news; Katsuno et al., 2010). They were surprised to find the double-transgenic mice died about 10 days earlier than mSOD1 mice. TGF-β1’s effects vary with its context, explained co-author Tony Wyss-Coray of Stanford University, California. While neuronal TGF-β1 protects nerves, astrocyte TGF-β1 seems to be damaging, he said.
How did astrocyte TGF-β1 make the animals worse? To find out, Endo analyzed the mRNA in the spinal cord. Compared with mSOD1 mice, the double-transgenics produced lower levels of microglial activation markers, such as CD68, and less of the neuroprotective proteins made by microglia, including insulin-like growth factor 1. This implied that TGF-β1 from astrocytes dampened microglial activation. Indeed, conditioned media from cultured mSOD1 astrocytes that overproduced TGF-β1 caused microglia to shrink and express fewer activation markers. Endo and Yamanaka did not confirm that TGF-β1 itself, rather than some other factor downstream of it, was responsible for making the microglia quiescent. Even so, they concluded that the astrocytes directly influenced microglia to be less neuroprotective.
However, Endo also discovered that the astrocytes acted on microglia indirectly, via T cells. In the mRNA data, he saw that T cell cytokines, such as IL-4, were below normal levels in the double-transgenics. T cells secrete these microglia-activating cytokines when they infiltrate the spinal cord in response to stress. Flow cytometry experiments showed fewer T cells in the cords of the double-transgenics. Those T cells that did make it in expressed more interferon-γ than interleukin-4, a profile associated with microglial deactivation. From its direct and indirect effects, astrocytic TGF-β1 lulls microglia into a quiescent state, dampening their production of neurotrophic factors, the authors concluded (see model below).
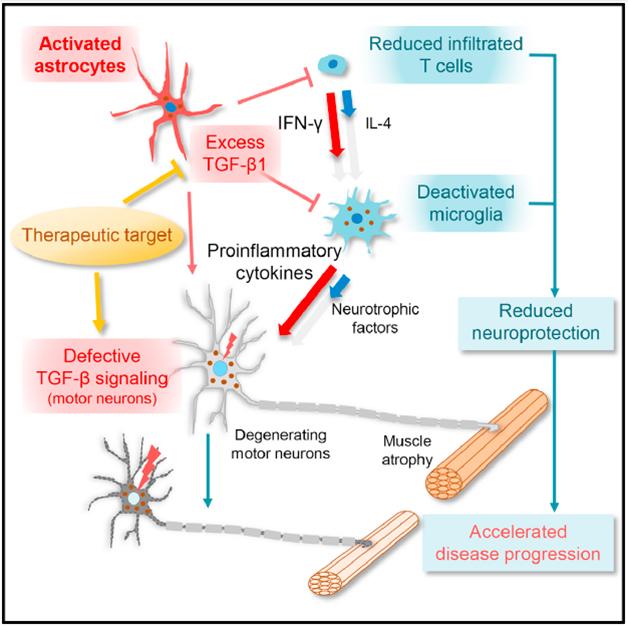
Bad neighborhood: In the spinal cords of ALS mice, TGF-β1 from astrocytes deactivates microglia, spoiling the environment for motor neurons. [Image courtesy of Cell Reports, Endo et al.]
TGF-β1 partly, but not fully, explains how astrocytes attack motor neurons, commented Isaac Chiu of Harvard Medical School. “This paper highlights how the milieu of the neurons is important,” he said. Hemali Phatnani of the New York Genome Center agreed. “It is the entire microenvironment that is affected [in ALS], and we should be studying crosstalk not just between glia and neurons, but between the types of glial cells,” she said. Neither Chiu nor Phatnani were involved in the Cell Reports study.
To test whether blocking TGF-β1 could protect against motor neuron disease, Endo injected mSOD1 mice with the small-molecule TGF-β1 inhibitor SB-431542, starting at 112 days when they were already ill. The treated mice lived nine days longer than untreated animals. This experiment used only eight mice per group, and the effect was modest but promising, Chiu said. Both he and Phatnani wanted to see more data, such as immunostaining to indicate what was happening with the mice’s T cells and microglia. Yamanaka told Alzforum the lab is working to confirm that SB-431542 treatment affects microglia and T cells in the spinal cord. He speculated that people who naturally have low levels of TGF-β1 might survive longer with ALS, while those with high levels might one day be candidates for a TGF-β1-blocker. SB-431542 is not an active investigational drug.
The idea that TGF-β1, which dampens inflammation, could be damaging contradicts the conventional wisdom that neuroinflammation worsens neurodegenerative disease. “The old concept, that any inflammation is bad for you, is not holding water,” commented Terrence Town of the University of Southern California in Los Angeles, who was not involved in the work. “This paper extends the notion that there are beneficial forms of inflammation that you might not want to quash.” That may partly explain why anti-inflammatory drugs did not protect people in AD clinical trials, he said (see May 2008 news). "You really want to selectively dampen the neurotoxic portion,” said Town. The difficulty, of course, is to dissect out the desirable and the damaging portions of inflammation. “This has become the critical question in neurodegenerative disease,” Town said.—Amber Dance
References
News Citations
Paper Citations
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008 Mar;11(3):251-3. PubMed.
- Houi K, Kobayashi T, Kato S, Mochio S, Inoue K. Increased plasma TGF-beta1 in patients with amyotrophic lateral sclerosis. Acta Neurol Scand. 2002 Nov;106(5):299-301. PubMed.
- Iłzecka J, Stelmasiak Z, Dobosz B. Transforming growth factor-Beta 1 (tgf-Beta 1) in patients with amyotrophic lateral sclerosis. Cytokine. 2002 Dec 7;20(5):239-43. PubMed.
- Katsuno M, Adachi H, Minamiyama M, Waza M, Doi H, Kondo N, Mizoguchi H, Nitta A, Yamada K, Banno H, Suzuki K, Tanaka F, Sobue G. Disrupted transforming growth factor-beta signaling in spinal and bulbar muscular atrophy. J Neurosci. 2010 Apr 21;30(16):5702-12. PubMed.
External Citations
Further Reading
Papers
- Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O'Keeffe S, Phatnani HP, Muratet M, Carroll MC, Levy S, Tavazoie S, Myers RM, Maniatis T. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013 Jul 25;4(2):385-401. PubMed.
- Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006 Oct 5;52(1):39-59. PubMed.
- Chen JH, Ke KF, Lu JH, Qiu YH, Peng YP. Protection of TGF-β1 against neuroinflammation and neurodegeneration in Aβ1-42-induced Alzheimer's disease model rats. PLoS One. 2015;10(2):e0116549. Epub 2015 Feb 6 PubMed.
- Shen WX, Chen JH, Lu JH, Peng YP, Qiu YH. TGF-β1 protection against Aβ1-42-induced neuroinflammation and neurodegeneration in rats. Int J Mol Sci. 2014 Dec 1;15(12):22092-108. PubMed.
- Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer's disease, role of cytokines. ScientificWorldJournal. 2012;2012:756357. PubMed.
- Endo F, Yamanaka K. [Neuroinflammation in amyotrophic lateral sclerosis]. Rinsho Shinkeigaku. 2014;54(12):1128-31. PubMed.
- Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, Siklos L, McKercher SR, Appel SH. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006 Oct 24;103(43):16021-6. PubMed.
- Chiu IM, Chen A, Zheng Y, Kosaras B, Tsiftsoglou SA, Vartanian TK, Brown RH Jr, Carroll MC. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci U S A. 2008 Nov 18;105(46):17913-8. Epub 2008 Nov 7 PubMed.
- Phatnani HP, Guarnieri P, Friedman BA, Carrasco MA, Muratet M, O'Keeffe S, Nwakeze C, Pauli-Behn F, Newberry KM, Meadows SK, Tapia JC, Myers RM, Maniatis T. Intricate interplay between astrocytes and motor neurons in ALS. Proc Natl Acad Sci U S A. 2013 Feb 19;110(8):E756-65. PubMed.
News
- Glia—Absolving Neurons of Motor Neuron Disease
- ALS in a Dish? Studying Motor Neurons from Human Stem Cells
- Believe It: Astrocytes Kill Neurons in New ALS Model
- ALS: Many Disparate Diseases, or Just Two?
- Geriatric Astrocytes Fail Motor Neurons in ALS Model
- In ALS, Astrocytes Use Ion Pump to Blast Motor Neurons
- Microglia in ALS: Helpful, Harmful, or Neutral?
- Peripheral Innate Immunity—Not So Peripheral to ALS?
- ALS: T Cells Step Up
Primary Papers
- Endo F, Komine O, Fujimori-Tonou N, Katsuno M, Jin S, Watanabe S, Sobue G, Dezawa M, Wyss-Coray T, Yamanaka K. Astrocyte-derived TGF-β1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep. 2015 Apr 28;11(4):592-604. Epub 2015 Apr 16 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Michigan State University
I think the observations that increasing TGFβ shortens lifespan and blocking TGFβ increases lifespan (assuming the drug does what it is intended to do) are important. ALS has no effective treatments. The paper reinforces the idea that microglial activation can have both beneficial and detrimental influences on neurodegeneration. Not only is the "state/phenotype" of activation critical, so is the magnitude of activation. The T cell infiltration component is intriguing.
View all comments by Dave MorganWeizmann Institute of Science
It is becoming widely accepted that CNS-infiltrating immune cells play a key role in fighting neurodegenerative conditions. Nevertheless, in most chronic neurodegenerative diseases, the recruitment of these cells into the CNS seems to be insufficient or delayed, resulting in a neuroinflammatory response that contributes to the exacerbation of the pathology. Thus, the neuroinflammation in neurodegenerative diseases has a negative reputation. In this study by Endo and colleagues, the authors examined the involvement of astrocyte-produced TGF-β1 on ALS disease progression in the mSOD1 (G93A) mouse model. TGF-β1 is one of the key cytokines that maintains the “immune-privileged” status of the CNS, and as the authors show, it has a direct immunosuppressive effect on microglia. In addition, they show an indirect effect on microglial phenotype through TGF-β1effect on T cells. It seems that the TGF-β-dependent mechanisms operating in CNS maintenance under physiological conditions might become counterproductive and even destructive under chronic pathology. Accordingly, the authors show that astrocyte-specific TGF-β1 overexpression in the spinal cord of mSOD1 mice is associated with a change in the local microenvironment, including a shift toward an IFN--dominant milieu within the spinal cord, and decreased expression of a wide array of chemokines, which correlated with reduced T cell numbers, and exacerbation of disease progression.
The finding by Endo and colleagues, with respect to T cells in ALS, are consistent with previous studies, pointing to an intriguing inverse correlation between the number of CNS-infiltrating CD4+ T cells and ALS disease progression; augmented T cell recruitment to the CNS is associated with a neuroprotective effect, while T cell deficiency is associated with a worsening effect on pathology. In a study by our group, published last week (Kunis et al., 2015), we found that in the mSOD1 mouse model, recruitment of immunoregulatory cells to the spinal cord (both monocyte-derived macrophages and regulatory T cells) can shift the local milieu toward an anti-inflammatory response, associated with mitigation of pathology and increased lifespan of the mice. Interestingly, we found that the shift toward an anti-inflammatory milieu, subsequent to the recruitment of effector CD4+ T cells, was associated with an elevation in TGF-β1 in the spinal cord of the mSOD1 mice. The findings by Endo et al., together with our results, may suggest a time-dependent effect for the anti-inflammatory response within the CNS; if prematurely induced, it might block the recruitment of neuroprotective immune cells.
References:
Kunis G, Baruch K, Miller O, Schwartz M. Immunization with a Myelin-Derived Antigen Activates the Brain's Choroid Plexus for Recruitment of Immunoregulatory Cells to the CNS and Attenuates Disease Progression in a Mouse Model of ALS. J Neurosci. 2015 Apr 22;35(16):6381-93. PubMed.
View all comments by Michal SchwartzMake a Comment
To make a comment you must login or register.