Surprise: HDAC Goes Down, Not Up, in Alzheimer’s Disease
Quick Links
As gene regulation changes in Alzheimer’s disease, scientists believe that histone deacetylases (HDACs) go into overdrive, shutting down transcription of certain genes. Consequently, several research groups are exploring the potential of HDAC inhibitors as AD therapeutics, with at least two trials currently enrolling. The recent development of a PET tracer that recognizes class I HDACs in the brains of living people provides a valuable tool for such trials. Now, however, the first HDAC PET data from people with AD upends previous findings. At the Alzheimer’s Association International Conference in Chicago July 22–26, Tharick Pascoal of McGill University, Montreal, reported that HDAC levels drop as disease advances.
- First HDAC PET data from AD patients reveals stark drop.
- HDAC class I isoforms are low in regions where amyloid and tau are high.
- The data raise questions about HDAC inhibition in AD trials.
“That was a surprise to us. We expected the opposite result,” Pascoal told Alzforum. At first, the researchers worried that there might be errors in their methodology; however, validation by a second group studying an independent cohort convinced them the finding was real, and robust. “I’ve never seen two independent PET studies where the images were so similar,” Pascoal said.
The HDAC ligand, [11C]Martinostat, was developed by Jacob Hooker of Massachusetts General Hospital, Boston, in collaboration with other groups. The tracer enters the brain readily and binds the three class I histone deacetylases, HDAC1, 2, and 3 (Wang et al., 2014; Wey et al., 2015). In healthy young controls, the tracer lights up specific brain regions, with the strongest signal in the putamen and cerebellum and weaker signal in the hippocampus and amygdala. The same pattern occurs in all volunteers, suggesting tight regulation of HDAC expression (Aug 2016 news).
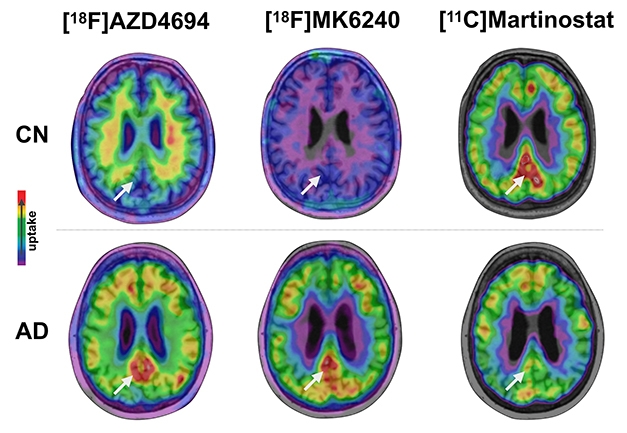
High AD Pathology, Low HDAC. In AD brains, amyloid plaques (left) and tau tangles (middle) accumulate while HDAC levels (right) wane in posterior cingulate cortices (arrow), compared with healthy elderly control brains. [Courtesy of Tharick Pascoal.]
Pascoal, working with Pedro Rosa-Neto and Serge Gauthier at McGill, used the tracer to scan 48 volunteers seen at McGill. Two of them were young healthy controls, 15 were cognitively healthy elderly, 15 had MCI, and 16 were diagnosed with AD. All also underwent amyloid scans with AZD4694, aka NAV4694 (May 2010 news), and tau imaging with MK6240. The cognitively healthy older participants were amyloid-negative.
In Chicago, Pascoal reported that brain regions that were positive for both plaques and tangles took up less Martinostat signal than control brains did. The affected regions— posterior cingulate cortex, precuneus, hippocampus, inferior parietal lobe, and lateral temporal lobe—all form part of the posterior default mode network, which accumulates pathology early in Alzheimer’s disease. The greater the AD pathology in these regions was in a person, the lower his or her Martinostat signal. The difference between cognitively healthy elderly and AD brains was stark, with almost no overlap between the two. In terms of SUVR, the Martinostat signal in young healthy controls was 1.5; in healthy elderly it ranged between 1.5 and 1.3; and in AD it ranged from just under 1.3 to 1. People with MCI had an intermediate level of Martinostat signal, suggesting that levels drop as disease develops, Pascoal said.
Pascoal and colleagues compared their data to a separate study done by Hooker and colleagues at the Martinos Center for Biomedical Imaging at MGH on 23 young people, 13 cognitively healthy elderly, and 10 AD patients. The two research groups used different protocols and analyses; even so, they saw the exact same HDAC pattern. “That gives the findings a lot of rigor,” Hooker told Alzforum. At AAIC, Pascoal presented pooled analyses from both cohorts.
What do low levels of HDAC in AD mean? Hooker noted the reduction in HDAC either could be compensatory or it could be part of disease pathology. A statistical analysis suggested the latter was more likely. Pascoal correlated regional HDAC, amyloid, and tau pathology to MMSE scores. He found that adding HDAC levels to the model increased its power to predict cognitive deficits. Amyloid and tau together explained about 70 percent of the sample’s variance in cognition, and with HDAC added, this rose to 90 percent. The data suggest that HDAC levels may mediate some of the damaging effects of Alzheimer’s pathology on cognition, Pascoal said.
Why has previous human research not seen this dramatic decrease in HDAC? Pascoal suggested this is because previous research was all postmortem and pathologists looked in the wrong regions. Most previous studies examined the medial temporal lobe or prefrontal cortex, where there is no change in Martinostat signal. The McGill group followed up on their own live imaging findings with a western blot analysis of postmortem samples from nine healthy elderly and six AD brains. That confirmed low levels of HDACs 1, 2, and 3 in AD brains in those same posterior DMN regions that had low Martinostat signal in the imaging study. “The take-home message from this research is that you have to be attentive to brain region,” Rosa-Neto told Alzforum. “The disease process is dynamic, and Alzheimer’s pathophysiology varies from region to region.”
In addition, much of the previous research on HDACs has been done in mouse models, which do not always reflect human biology. Several studies have reported less histone acetylation in the hippocampi of various neurodegenerative disease models, including APP/PS1 AD mice, than in wild-type, and HDAC inhibitors improved learning in these models (Jun 2004 news; May 2007 news; Francis et al., 2009; Kilgore et al., 2010; Mar 2012 news).
For their part, Pascoal and colleagues performed a western blot analysis of postmortem samples from two rat models of AD. The R-Thy1-APP line (Leon et al., 2010) carries a human amyloid transgene and does not develop neurofibrillary tangles; it showed no change in class I HDAC levels compared to wild-type rats of the same age, Pascoal said in Chicago. On the other hand, the TgF344 rat (Cohen et al., 2013) develops both amyloid plaques and tau tangles; it had lower HDAC1 and 2 levels than wild-type. The findings suggest that, just as in human brain regions, both amyloid and tau pathology are required to change HDAC levels, Pascoal said. Both findings contrast with mouse data showing increased HDAC in AD models, suggesting the mouse findings might not translate to other species.
There is another wrinkle, though. The Martinostat tracer does not distinguish between particular class I isoforms. Li-Huei Tsai of the Massachusetts Institute of Technology noted that some of these may move in different directions in AD. For example, Tsai has evidence that HDAC2 rises in the hippocampi of AD mice, whereas other isoforms, such as HDAC1, may fall. HDAC2 has been tied to harmful effects, while HDAC1 and a class III isoform, SIRT1, seem to be neuroprotective in mice (Dec 2008 conference news; May 2009 news; Feb 2010 conference news). “Concerning using HDAC inhibitors for treatment, obviously isoform specificity would be very important,” Tsai wrote to Alzforum. She did not hear Pascoal’s AAIC talk.
Hooker acknowledges that Martinostat’s lack of specificity is a limitation. Alas, he noted, available drugs don’t distinguish between class I isoforms, either. “We believe the pan-class Martinostat is useful to judge the overall HDAC, or epigenetic, ‘tone,’” Hooker told Alzforum. Another potential limitation of the tracer is that it measures HDAC protein level, not necessarily its activation state. “We assume that HDAC density and activity are linked, but HDAC [regulation] is complicated. As with all PET imaging agents in humans, we have to be cautious with the exact interpretation,” Hooker noted.
How does this information affect HDAC inhibitor trials? The McGill group recommended caution. “Because HDAC class I levels are already low in AD, there is the potential that reducing them further could cause harm,” Pascoal said.
A Phase 1 open-label dose-finding study of the HDAC inhibitor vorinostat is currently enrolling in Germany. Vorinostat inhibits class I and class II HDACs. The McGill group has shared their data with the German investigators so they can take it into account. Notably, the class I HDAC inhibitor valproate was previously found ineffective for managing neuropsychiatric symptoms in AD, and it also associated with worse cognitive decline (Fleisher et al., 2011).
Meanwhile, researchers at the University of California, Irvine, are investigating the class III HDAC inhibitor nicotinamide for AD. They wrapped up a Phase 1 safety study of 50 AD patients in 2014, and are currently enrolling for Phase 2. The Martinostat findings provide no information on class III HDACs, Pascoal noted.—Madolyn Bowman Rogers
References
News Citations
- Epigenetic Tracer Uncovers Patterns of Healthy Gene Regulation
- Geneva: The AstraZeneca Ligand—The Fairest of Them All?
- For Better Memory, Try Keeping Your HAT On…
- Memories—Forgotten, But Not Gone?
- Does Epigenetic Modification by Aβ Offer New Take on Therapy?
- Overworked HDACs Leave Transcriptional Posts to Push DNA Repair
- It’s an HDAC2 Wrap— Memory-suppressing DNA Modifier Identified
- Copper Mountain: Knight Vision—SIRT1 Aids ADAM10, Slays Aβ
Therapeutics Citations
Paper Citations
- Wang C, Schroeder FA, Wey HY, Borra R, Wagner FF, Reis S, Kim SW, Holson EB, Haggarty SJ, Hooker JM. In vivo imaging of histone deacetylases (HDACs) in the central nervous system and major peripheral organs. J Med Chem. 2014 Oct 9;57(19):7999-8009. Epub 2014 Sep 18 PubMed.
- Wey HY, Wang C, Schroeder FA, Logan J, Price JC, Hooker JM. Kinetic Analysis and Quantification of [¹¹C]Martinostat for in Vivo HDAC Imaging of the Brain. ACS Chem Neurosci. 2015 May 20;6(5):708-15. Epub 2015 Mar 25 PubMed.
- Francis YI, Fà M, Ashraf H, Zhang H, Staniszewski A, Latchman DS, Arancio O. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer's disease. J Alzheimers Dis. 2009;18(1):131-9. PubMed.
- Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, Rumbaugh G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2010 Mar;35(4):870-80. PubMed.
- Leon WC, Canneva F, Partridge V, Allard S, Ferretti MT, DeWilde A, Vercauteren F, Atifeh R, Ducatenzeiler A, Klein W, Szyf M, Alhonen L, Cuello AC. A novel transgenic rat model with a full Alzheimer's-like amyloid pathology displays pre-plaque intracellular amyloid-beta-associated cognitive impairment. J Alzheimers Dis. 2010;20(1):113-26. PubMed.
- Cohen RM, Rezai-Zadeh K, Weitz TM, Rentsendorj A, Gate D, Spivak I, Bholat Y, Vasilevko V, Glabe CG, Breunig JJ, Rakic P, Davtyan H, Agadjanyan MG, Kepe V, Barrio JR, Bannykh S, Szekely CA, Pechnick RN, Town T. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric aβ, and frank neuronal loss. J Neurosci. 2013 Apr 10;33(15):6245-56. PubMed.
- Fleisher AS, Truran D, Mai JT, Langbaum JB, Aisen PS, Cummings JL, Jack CR, Weiner MW, Thomas RG, Schneider LS, Tariot PN, . Chronic divalproex sodium use and brain atrophy in Alzheimer disease. Neurology. 2011 Sep 27;77(13):1263-71. PubMed.
External Citations
Further Reading
News
- It’s All in the Packaging: Better Delivery Renders HDAC Combo Effective in Mice
- Progranulin-Boosting Drug Moves into Phase 2 for Frontotemporal Dementia
- Does Epigenetic Modification by Aβ Offer New Take on Therapy?
- Histone Acetylation: Epigenetic Achilles’ Heel of Memory in Aging
- Only in Old Mice, a Touch of Cannabinoid Helps Memory
Annotate
To make an annotation you must Login or Register.
Comments
The University of Auckland
These findings complement our previous work in this area (Narayan et al., 2015), raising concern about the use of HDAC inhibitors in the context of AD. The reduction in HDACs observed independently by Pascoal et al. and Hooker et al. explains some of the changes we have observed in global histone acetylation levels. We have found significant increases in these modifications in postmortem AD brains in the inferior temporal gyrus and middle temporal gyrus.
Also of great interest is that these independent groups have associated reduction in HDAC levels with AD-related pathology (plaques and tangles). We, too, found significant association between these pathological markers and histone acetylation levels; however, the histone acetylation changes correlated significantly with changes in total histone protein levels, and our data also reflected a substantial compromise in protein degradation pathways underlying these changes.…More
References:
Narayan PJ, Lill C, Faull R, Curtis MA, Dragunow M. Increased acetyl and total histone levels in post-mortem Alzheimer's disease brain. Neurobiol Dis. 2015 Feb;74:281-94. Epub 2014 Dec 5 PubMed.
University of Miami
These [11C]Martinostat PET studies are nicely executed, informative, and potentially useful for future CNS drug-discovery pursuits when studying target engagement, etc.
However, they do not help validate/invalidate HDACs as drug targets for AD or any other indication. Epigenetic phenomena, in this case acetylation, affecting the structure of histones (and many other proteins), result from the actions of many effectors. The HDACs, a subclass of which is being labeled by [11C]Martinostat, are just a few among hundreds of proteins that affect chromatin structure through balancing mechanisms. What matters functionally is the net effect on any modification, here acetylation, in a given context, and its consequences on gene expression and a myriad of other cellular effects. Needless to say, there will of course never exist PET ligands for all these various effectors to study them simultaneously.…More
Finally, while not proposing that HDAC inhibition would be truly disease-modifying in AD, we and others have over the years been struck by the ability of HDAC inhibitors to remedy AD hallmarks, and improve cognition, in a wide variety of models. We feel that existing synthetic and natural HDAC inhibitors, and ones optimized for kinetics, specificity, safety, etc., deserve close attention by the AD field.
Make a Comment
To make a comment you must login or register.