SOD1, Tau Swept Up in Prion-like Biology at ALS Meeting
Quick Links
The first outward sign of amyotrophic lateral sclerosis (ALS) usually crops up in one spot: a little trouble swallowing, or weakness in one leg or one arm. Then it spreads. Scientists now suspect the underlying pathogenic proteins, such as SOD1, might pave the way for this symptom spread in prion-like fashion, traveling from neuron to neuron and converting good protein to bad. This was a major theme at the International Symposium on ALS/MND held December 11-13 in Orlando, Florida. Adopting injection seeding paradigms that have long been used in prion research, researchers find parallels between the structural variants of proteins involved in neurodegeneration, such as SOD1 and tau, and the “strains” of varying pathogenicity in prion biology. Importantly, scientists were not going so far as to call ALS a prion disease, according to Brian Dickie of the Motor Neurone Disease Association in the United Kingdom, which organized the conference. All the same, if ALS is driven by a prion-like mechanism, then treatments should be able to halt its progression, he speculated. Some speakers offered preliminary ideas about how to find and test such therapeutics.
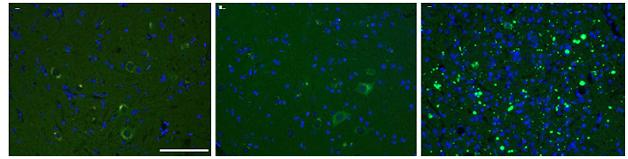
Pass it on. SOD1 aggregates in mice injected with tissue homogenates containing the SOD1-G93A “strain” (right), but not buffer (left) or homogenates from SOD1-G85R mice (center). [Courtesy of Acta Neuropathologica/Springer (Jan 2016, Vol 131, Issue 1, pp 103-114).]
SOD1 Gets a Move On
Jacob Ayers of the University of Florida in Gainesville described strain-like properties of SOD1 he discovered during his efforts to follow SOD1 transmission in vivo (Ayers et al., 2014). As models, he used mice heterozygous for the ALS-associated human SOD1 variant G85R. He tracked a version of SOD1 G85R fused to yellow fluorescent protein (YFP) to see if the protein aggregated in vivo (see Jan 2009 news). These heterozygous mice do not develop motor neuron disease on their own. However, their genotype somehow made them vulnerable to the spread of toxic forms of SOD triggered by seeding injection experiments, compared to mice expressing only wild-type SOD1.
Ayers injected newborn mice intrathecally with spinal cord homogenate from paralyzed adult mice expressing toxic SOD1-G93A or G37R, two other ALS-linked variants. Six of 10 G85R mice injected with SOD1-G93A developed paralysis in their hind limbs between three and 11 months of age. Animal autopsy, aka necropsy, revealed punctate yellow SOD1 aggregates in the neurons of the spinal cord, brainstem, and thalamus. When Ayers injected homogenates from these animals into four new SOD1-G85R-YFP mice, disease occurred even sooner, with all animals developing movement difficulties within three months. In contrast, non-transgenic mice, or mice expressing wild-type human SOD1, that were injected with SOD1-G93A homogenates appeared normal, as did G85R-YFP animals injected with homogenates from non-transgenic mice.
The findings indicate that passaging toxic SOD1 variants through animals expressing G85R can convert this otherwise mild variant into a more pathogenic strain. That strain then more readily corrupts additional molecules of G85R SOD1.
This accelerated transmission in the second passage mimics the phenomenon of host adaptation seen in prion biology, in which the disease transmits more quickly between proteins identical in sequence, Ayers said (Aguzzi et al., 2007; Bruce et al., 1994). With prions, the concept usually refers to transfer between animals of the same versus different species; in this case it relates to the SOD1 genotype of the mice.
Ayers saw different results with the G37R and the G93A SOD1 variant. Even after more than 13 months, SOD1-G85R-YFP injected with homogenates from G37R animals developed no signs of motor neuron disease. In prion terms, SOD1-G93A and G37R would be considered different strains, Ayers pointed out.
In a study published last month, Ayers reported how SOD1 misfolding spread throughout the mouse’s nervous system following injection into the hind limb sciatic nerves of adult mice. In this case, he first injected homogenates from G93A mice intrathecally into SOD1-G85R heterozygotes. When these animals became paralyzed he used their tissue to make new homogenates that he injected directly into the sciatic nerves of a new batch of G85R mice. Aggregates appeared first in sensory neurons in the dorsal root ganglion on the injected side of the body, and from there spread to the contralateral dorsal root ganglion, spinal cord, and brain. In parallel, the mice lost strength first in the injected leg, then in the other. Within a month of the injection, both hind legs were paralyzed (Ayers et al., 2016).
The results represent a “grand slam,” commented Neil Cashman of the University of British Columbia in Vancouver, Canada, who did not participate in the project. “They have been able to develop a credible, in vivo model of SOD1-propagated misfolding, accompanied by paralysis,” Cashman said. SOD1-G85 seems to act as a sort of “acceptor” for SOD1-propagated misfolding in a way that the wild-type protein does not. Cashman speculated that in an aging person, wild-type SOD1 might become more prone to templated misfolding over time. This might be due to changes in proteostasis that leave it incompletely folded, or as a monomer instead of its natural dimer form, he theorized. This propagation contrasts the lack of pathology spread in other ALS models based on ubiquitous SOD1 transgene expression, Cashman noted. Ayers suggested that scientists could test therapeutics in mice injected with second-passage homogenates.
Edward Pokrishevsky, in Cashman’s group, also had therapeutics in mind. Pokrishevsky developed a high-throughput assay, based on the principles of toxic protein propagation described by Ayers, and used it to screen for possible blockers of SOD1 misfolding and propagation. In HEK293 cells, Pokrishevsky co-expressed SOD1-G85R tagged with green fluorescent protein (GFP), plus another more-toxic version of SOD1, such as the truncation mutant G127X. Alone, SOD1-G85R-GFP appeared diffuse, but in the presence of the second toxic SOD1 it aggregated in 20 percent to 30 percent of cells. Pokrishevsky reasoned that drugs that prevent SOD1 propagation should protect SOD1-G85R from aggregation, and could thus be identified in his assay
The researchers have not yet started a large screen, but Pokrishevsky presented uridine as a test case. He became interested in it because 5-fluorouridine (5-FU) binds to SOD1 at a tryptophan residue crucial for templated misfolding, suggesting it might block the conversion of new SOD1 molecules (see Oct 2011 news; Dec 2012 conference news; Wright et al., 2012). The Cashman lab found that 5-FU did indeed act in this manner. However, 5-FU is not a good therapeutic option for ALS because it can kill cells, said Pokrishevsky. It replaces uracil in DNA and RNA, blocking DNA and RNA synthesis, and also inhibits thymidylate synthase, disrupting production of thymine.
Because uridine has been used in people to counteract 5-FU toxicity, Pokrishevsky tested both together. The combination prevented SOD1-G85R-GFP aggregation. Surprisingly, so did uridine alone. Might uridine be a better drug candidate? Unfortunately, the dose needed was so high as to be impractical in vivo. Cashman told Alzforum his group plans to screen more molecules with Pokrishevsky’s assay this year.
The assay depends on mutant SOD1 to start and maintain propagation, but in other presentations Pokrishevsky and Cashman explained how wild-type SOD1 might also propagate as a prion. This may be important because SOD1 aggregates turn up in sporadic cases and in ALS caused by other mutations (see Oct 2010 news; Forsberg et al., 2010). The researchers wondered if variants of the ALS genes TDP-43 and FUS, or the cytoplasmic mislocalization of TDP-43 protein that marks nearly all human ALS, might lead to SOD1 misfolding and propagation. “Perhaps misfolded SOD1 is the common factor across all ALS,” Pokrishevsky told Alzforum.
If so, then conditioned media from HEK cultures expressing ALS-linked versions of TDP-43 or FUS ought to contain misfolded SOD1 that transmits its conformation to fresh cell cultures. Indeed, Pokrishevsky found that an antibody specific for misfolded SOD1 detected antigen in fresh recipient cells—either HEK cells or primary mouse neurons—but not if the antibody for misfolded SOD1 was first added to the conditioned media. Cashman concluded that once SOD1 has misfolded in one group of cells, it acquires the ability to transmit its altered conformation from cell to cell, like a prion. The lab is gearing up to test this idea in Pokrishevsky’s aggregation assay, Cashman told Alzforum. Notably, TDP-43 and FUS proteins in the recipient cells stayed where they were; they did not have to mislocalize or aggregate to support further SOD1 misfolding once it had started.
Ayers called the finding provocative, and noted it fits with Cashman’s previous work linking TDP-43 pathology and SOD1 misfolding in people with sporadic ALS (Pokrishevsky et al., 2012). Cashman speculated that treatment with the antibody to misfolded SOD1, or a therapy that knocks down normal SOD1 expression, could slow or halt ALS progression.
The Diversity of Tau
Marc Diamond of the University of Texas Southwestern Medical Center in Dallas discussed different strains of tau. He noted that despite being based on just one protein, tauopathies manifest in myriad ways, ranging from Alzheimer’s and frontotemporal dementias and chronic traumatic encephalopathy to progressive supranuclear palsy and corticobasal degeneration. Different strains could explain both the progression of tau pathology across the brain and the diverse phenotypes, he reasoned.
In 2014, Diamond and colleagues reported their isolation of many different strains of tau. Studying two, they found that one produced large, cytoplasmic aggregates in cells and neurofibrillary tangles in mice. The other formed small, nuclear inclusions both in culture and the hippocampus. Importantly, as the researchers transferred brain material from affected to new mice, each strain maintained its phenotype (see May 2014 news).
In Orlando, Diamond reported his lab’s progress since then with similar characterization of 18 different strains of tau aggregates: eight from patient tissues, two from tau-P301S transgenic mice, and eight from recombinant tau fibrillized in vitro. The lab propagates them in cell cultures expressing tau. Since long-term expression of full-length tau can damage dividing cells, they use a HEK293 line expressing only the core part of tau that allows aggregation, amino acids 244-372, harboring V337M and P301L mutations and fused to YFP. The scientists originally chose this cell line because they thought it would be ideal to detect tau aggregates, Diamond told Alzforum. Disease-linked tau mutations usually make the protein more susceptible to prion conversion, he said, although wild-type tau can also be converted at a lower rate.
The strains had distinct identities. In cell culture, most were quite stable, displaying characteristic aggregates such as threadlike fibrils or punctate inclusions. Some aggregate types killed all the cells in a culture, while others killed only a subset. The deadliest strains were also the best at seeding new aggregates in cultured cells, Diamond said.
To study the structure of the strains, the researchers borrowed a technique from the prion world, using a protease to digest each strain. The protease completely degraded normal tau, but the aggregating strains yielded distinctive patterns of fragmentation depending on which portions of their structure were protected from digestion (see Aug 2013 conference news). These digestion patterns did not correlate in any way with toxicity or seeding ability, Diamond told Alzforum.
To learn what these tau strains might do in the brain, the scientists injected them into the hippocampi of transgenic mice expressing tau-P301S. They saw different patterns of pathology. For example, one strain remained in the hippocampus while another spread into the cortex. Some strains seeded widespread aggregation of endogenous tau within four weeks; others took a couple of months. The researchers noticed that the brain pathology did correlate with the protease digestion patterns, in that strains with similar digestion patterns had similar in vivo distributions.
Kevin Talbot of Oxford University in the United Kingdom, who led the meeting’s program committee, praised Diamond’s work, saying the different strains could explain how the same protein causes different diseases.
Unlike Ayers’ SOD1 mice, Diamond’s injected mice did not get sick within the two months the researchers have observed them thus far, he told Alzforum. He will monitor behavior and survival over longer time periods.
Dickie speculated that if ALS proteins do behave like prions, then future treatments could put up a sort of firewall to keep the problem isolated. In theory, this might limit symptoms to one body part and slow progression. Small molecules or antibodies might block the transit of prion-like proteins, Diamond suggested.—Amber Dance
References
News Citations
- Bad Origami: Misfolded SOD1 in Living Color
- ALS: Sticky Tryptophan Achilles' Heel for Superoxide Dismutase?
- Chicago—ALS Protein SOD1 Painted as Disease Template
- Research Brief: SOD1 in Sporadic ALS Suggests Common Pathway
- Like Prions, Tau Strains Are True to Form
- Are Protein Strains The Cause of Different Tauopathies?
Paper Citations
- Ayers JI, Fromholt S, Koch M, DeBosier A, McMahon B, Xu G, Borchelt DR. Experimental transmissibility of mutant SOD1 motor neuron disease. Acta Neuropathol. 2014 Dec;128(6):791-803. Epub 2014 Sep 28 PubMed.
- Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007 Jul;8(7):552-61. PubMed.
- Bruce M, Chree A, McConnell I, Foster J, Pearson G, Fraser H. Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Philos Trans R Soc Lond B Biol Sci. 1994 Mar 29;343(1306):405-11. PubMed.
- Ayers JI, Fromholt SE, O'Neal VM, Diamond JH, Borchelt DR. Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathol. 2016 Jan;131(1):103-14. Epub 2015 Dec 9 PubMed.
- Wright GS, Antonyuk SV, Kershaw NM, Strange RW, Samar Hasnain S. Ligand binding and aggregation of pathogenic SOD1. Nat Commun. 2013;4:1758. PubMed.
- Forsberg K, Jonsson PA, Andersen PM, Bergemalm D, Graffmo KS, Hultdin M, Jacobsson J, Rosquist R, Marklund SL, Brännström T. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS One. 2010;5(7):e11552. PubMed.
- Pokrishevsky E, Grad LI, Yousefi M, Wang J, Mackenzie IR, Cashman NR. Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS One. 2012;7(4):e35050. PubMed.
Further Reading
Papers
- Kanouchi T, Ohkubo T, Yokota T. Can regional spreading of amyotrophic lateral sclerosis motor symptoms be explained by prion-like propagation?. J Neurol Neurosurg Psychiatry. 2012 Jul;83(7):739-45. PubMed.
- Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology. 2009 Sep 8;73(10):805-11. PubMed.
- Ravits J. Focality, stochasticity and neuroanatomic propagation in ALS pathogenesis. Exp Neurol. 2014 Dec;262 Pt B:121-6. Epub 2014 Aug 6 PubMed.
- Grad LI, Yerbury JJ, Turner BJ, Guest WC, Pokrishevsky E, O'Neill MA, Yanai A, Silverman JM, Zeineddine R, Corcoran L, Kumita JR, Luheshi LM, Yousefi M, Coleman BM, Hill AF, Plotkin SS, Mackenzie IR, Cashman NR. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc Natl Acad Sci U S A. 2014 Mar 4;111(9):3620-5. Epub 2014 Feb 18 PubMed.
- Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, Kotzbauer PT, Miller TM, Papy-Garcia D, Diamond MI. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013 Aug 13;110(33):E3138-47. Epub 2013 Jul 29 PubMed.
- Ravits J, Paul P, Jorg C. Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology. 2007 May 8;68(19):1571-5. PubMed.
News
- Proteopathic Seeds and Neurodegenerative Diseases
- Tales of Traveling Tau: Is Transfer Between Neurons Normal?
- Tau Triple Threat: Do Trimers Make Bad Seeds?
- Cellular Biosensor Detects Tau Seeds Long Before They Sprout Pathology
- Do Tau "Prions" Lead the Way From Concussions to Progression?
- Alzheimer’s Transmission Between People? Amyloid Plaques in Hormone Recipients Hint at Prion-like Spread
- Prion Stabilizers Boost Survival in Infected Mice—What About Other Proteinopathies?
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.