Dysregulated Lipid Metabolism Comes to the Fore at AD/PD
Quick Links
After years of languishing in obscurity, the topic of disruptions in the brain's lipid metabolism is moving center stage. The number of publications in this area is growing, and at the International Conference on Alzheimer’s and Parkinson’s Diseases, held March 28 to April 1 in Gothenburg, Sweden, lipid dysfunction debuted as a session topic. At the session, six presenters showed off a tool kit of newfangled imaging and analysis methods that promise to transform this famously complex field of research. Their data converged on changes in phospholipid, fatty acid, and cholesterol metabolism in AD, PD, and Lewy body dementia. It appears that these perturbations occur early in the disease trajectory, perhaps even preceding amyloid plaques.
- Changes in phosphatidylcholine metabolism track with cognitive decline in AD.
- People with PD had more metabolites of this lipid in their blood.
- Lipids in the brain hit the skids prior to amyloid plaques.
- APOE4 oligodendrocytes and astrocytes contain excess cholesterol, other lipids.
“I think the session was excellent and really shows the coming of age of how we understand and measure lipid changes in Alzheimer’s disease (and Parkinson’s disease),” Rik van der Kant, Vrije Universiteit, Amsterdam, who did not speak at this session, wrote to Alzforum.
Ole Isacson of Harvard Medical School, Boston, co-chaired the session. “This [work] provides fundamental understanding for how these diseases can be caused by lipid dyshomeostasis and metabolic disturbances that drive downstream amyloidogenic pathways. Lipid dyshomeostasis is more of a driver than perhaps we have had time to investigate,” he told Alzforum. Laura Beth McIntire of Weill Cornell Medical College, New York City, welcomed the new focus on advances in lipidomics in neurodegenerative disease. “Lipid metabolism is a major pathway in AD,” she said.
Already in 1906, Alois Alzheimer saw glial cells that appeared bloated with lipid droplets under the microscope. Much later, human genetics pointed to lipid metabolism in neurodegenerative diseases. Variants of the lipid binding proteins ApoE and PICALM, the lipid transporter ABCA7, the genes MS4A4A and PLCG2, and others have been linked to higher odds of AD, as well as variants in the glycolipid hydrolase GBA1 to LBD and PD (Feb 2021 news; Apr 2018 news; Mar 2021 news). Up to 40 percent of AD genome-wide association study hits are involved in lipid metabolism, making it the major contributor to disease risk, McIntire noted.
To understand how lipid metabolism goes awry in AD, McIntire analyzed gene expression in cortical postmortem tissue samples from 639 participants from the Religious Orders Study and the Rush Memory and Aging Project (ROSMAP) cohort. After accounting for amyloid plaque and tau tangle loads, she found that people whose cognition slipped fastest had upregulated phospholipase A2 (PLA2) and downregulated lysophosphatidylcholine acyltransferase 2 (LPCAT2) genes. The former converts the lipid phosphatidylcholine (PC) to lysoPC; the latter recycles lysoPC back to PC in what is called the Lands cycle or acyl chain remodeling, a core constituent of the making and breaking of the phospholipids that form cell membranes (see image below). These and other gene-expression changes suggest that an abundance of lysoPC might reflect lipid metabolism dysregulation in AD, McIntire concluded at AD/PD.

Lipid Flux. To recycle and replenish phospholipids within cell membranes, phospholipase A2 (PLA2) cleaves a fatty acid tail from the lipid to make a lysophospholipid. A lysophospholipid acyltransferase, such as LPCAT2, tacks on a new tail to make a new phospholipid. The headgroup (X) distinguishes lipid subtypes, with phosphatidylcholine sporting a choline molecule. [Courtesy of Bankaitis, 2009, Journal of Cell Biology.]
Next, the scientists performed lipidomics on samples from 99 ROSMAP participants. Limitations in their lipid panel precluded them from defining which species were up- or downregulated in AD, but even so, they were able to analyze patterns in lipid abundance based on participants' clinical diagnosis and APOE4 genotype. Two profiles emerged. Lipidomes were similar in cognitively normal APOE4 carriers and all people with mild cognitive impairment, yet were significantly different from cognitively normal E4 noncarriers and all people who had AD. “If we’re only looking at advanced AD compared to cognitively normal people, we may be missing important lipid changes,” McIntire concluded.
To McIntire, this implies lipid metabolism changes so early in AD that they would precede amyloid plaques and neurofibrillary tangle deposition. Indeed, in ApoE4 carriers, PET imaging with a tracer binding the fatty acid DHA reflects a DHA deficiency in gray matter as early as age 35 (Yassine et al., 2017).
DHA levels change in young amyloidosis mice, too. Artur Lazarian, who works in McIntire’s lab, analyzed brain slices from wild-type and Tg2576 mice using imaging mass spectrometry. This technique ionizes molecules from a specific area within a brain tissue slice, measures the mass and abundance of each ion there, and creates maps of how each lipid species populates the brain. Incidentally, Lazarian mentioned that his lab has a new imaging mass spectrometer and invited collaboration inquiries from scientists interested in studying lipidomics.
Compared to wild-type mice, the transgenics accumulated more polyunsaturated fatty acids, including DHA, in the brain by 4 months of age, before plaques appeared. These lipids remained high and unchanged while amyloid accumulated. How would DHA, or other fatty acids and lipids, influence subsequent amyloid accumulation? “The role of lipids in amyloid pathology is potentially important, though still enigmatic,” Jörg Hanrieder, University of Gothenburg and University College London, wrote to Alzforum.
McIntire offered one explanation. It appears that certain types of fatty acid stick to Aβ and influence its accumulation. Most of the lipids at different abundances in the ROSMAP profiles contained polyunsaturated fatty acids. In an in vitro assay, McIntire found that the polyunsaturated DHA, but not the saturated stearic acid, bound fluorescently labeled Aβ42. Nuclear magnetic resonance and molecular modeling detected DHA docking into a pocket on Aβ42 created by residues 12 through 20. Thioflavin T aggregation assays showed that DHA prevented Aβ42 aggregation while stearic acid did not, likely because the latter did not bind to Aβ42.
This finding implies a caution for mechanistic studies using Aβ. “Aβ is probably characteristically lipidated, and it is unlikely that studies using the unlipidated peptide give us changes that are biologically relevant,” McIntire said. Van der Kant agreed. “I don’t think it would be farfetched to hypothesize that adding unlipidated synthetic Aβ to cells or mouse brains would lead to toxicity simply because this ‘empty’ Aβ starts searching for lipids to bind and extract,” he wrote to Alzforum.
To get a better look at how lipids and plaques interact in the brain, Hanrieder’s group has been developing a sophisticated method that uses imaging mass spectrometry to detect simultaneously which lipids are found in amyloid plaques and where they are within a given plaque. Hanrieder has studied brain tissue slices from Tg-ArcSwe mice (Kaya et al., 2017; Kaya et al., 2017; Michno et al., 2018). The scientists sharpened the 10-micron spatial resolution of their imaging MS down to 300 nanometers by combining it with hyperspectral confocal microscopy, which uses two different structurally sensitive fluorescent amyloid dyes to label diffuse or cored plaques, respectively (Wehrli et al., 2023). In essence, this multimodal analysis renders specific chemical information from mass spec at the high spatial resolution of amyloid microscopy, Hanrieder explained.
Hanrieder is using it on the human cortex. He started with postmortem samples from five people with familial AD and a presenilin 1 mutation, and is now analyzing sporadic AD tissue, from UCL's Queen Square Brain Bank. He was able to “see” phospholipids, ceramides, cerebrosides, and gangliosides—importantly, all of them localized to amyloid plaques.
One ganglioside, GM1, was the main lipid in cored plaques. In contrast, sulfatide, a major lipid component of myelin sheaths, was relatively absent from plaques and the surrounding areas (see image below). Hanrieder thinks that this suggests demyelination of nearby neurons.

Gangliosides, No Sulfatides. In presenilin 1 ADAD mutation carriers, amyloid plaques (green, left) include the ganglioside GM1 (green, middle) but lack a sulfatide species (blue, right). [Courtesy of Jörg Hanrieder, University of Gothenburg.]
At AD/PD, Li-Huei Tsai of Massachusetts Institute of Technology, Boston, also presented on disrupted myelination in APOE4 carriers with AD. Tsai's lab recently published results of single-nucleus RNA sequencing on cortical tissue from 32 ROSMAP participants, comparing gene expression between APOE genotypes. Relative to E3/3 carriers, most of the differentially expressed genes in E4/4 carriers mapped to lipid metabolism pathways, with myelination down- and cholesterol synthesis upregulated in oligodendrocytes. Cortical tissue from APOE3/4 carriers was reported to be less myelinated than in E3/3 carriers, and cultured iPSC-derived human APOE4/4 oligodendrocytes to contain more lipid droplets than their E3/3 counterparts. Tsai concluded that ApoE4’s impaired ability to transport the lipid cholesterol might hobble oligodendrocytes' ability to properly myelinate axons (Blanchard et al., 2022). Van der Kant noted that, somehow, AD risk variants seem to cause a broken feedback loop, where cholesterol accumulates intracellularly, yet cells keep on producing more cholesterol as if they were starved of the lipid.
Another AD GWAS hit, the lipid efflux pump ABCA7, reared its head here, too. Much like APOE4 carriers, 12 ROSMAP participants with a loss-of-function mutation in ABCA7 had upregulated cholesterol synthesis genes in their oligodendrocytes, according to Djuna Von Maydell from the Tsai lab. Oddly, their lipid metabolism was downregulated in other cells. Excitatory neurons expressed the most ABCA7, yet those harboring the AD risk variant downregulated genes involved in lipid metabolism. Lipidomics analysis showed that ABCA7 loss-of-function brains contained less of almost every lipid species, including phosphatidylcholine, than did noncarriers. Van der Kant wondered if cholesterol metabolism was abnormal in people with mutations in other AD-linked genes. “[I believe it] will likely be a key aspect of more AD risk variants,” he wrote.
What About Dementia with Lewy Bodies and Parkinson's?
Scientists have been implicating lipid dysregulation in LBD and PD for longer than in AD. Besides driving up risk of AD, APOE4 also puts a person at greater risk of developing Lewy body dementia. Mutations in the glucocerebrosidase GBA1 increase risk for LBD and Parkinson’s. GBA1 hydrolyzes glucocerebroside, an intermediate metabolite needed to make the glycolipids that are a component of cell membranes. At AD/PD, Isacson showed that blocking GBA1 in wild-type mice increased the amount of ApoE protein in their brain cortices and hippocampi, suggesting that ApoE adapts to the presence of more lipids when GBA1 no longer hydrolyzes them.
To explore how GBA1 and APOE influence brain lipid homeostasis, Isacson and colleagues treated wild-type and APOE knockout mice with the GBA1 inhibitor conduritol b epoxide for 18 days. In hippocampal tissue from both mouse lines, lots of lipids—cholesterol, glycolipids, triglycerides—formed large clusters between brain cells (see image below). Microglia multiplied, and levels of the complement activator C1q and the intracellular cholesterol trafficking protein NPC1 rose.
APOE knockouts had more severe phenotypes than did wild-types, likely because ApoE was not there to compensate for the lipid influx. “GBA1 inhibition causing glycolipid accumulation is functionally connected to the loss of APOE function causing cholesterol and lipid accumulation,” Isacson told Alzforum.
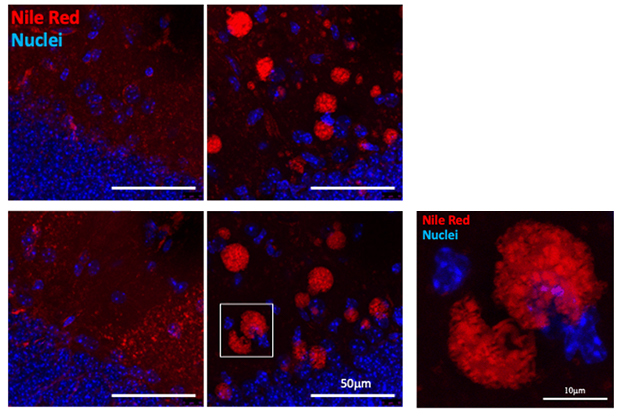
Little Balls of Fat. In the hippocampi of wild-type (top) or APOE knockout mice (bottom), lipids (red) formed blobs between brain cells when GBA1 was inhibited (right; nuclei in blue). [Courtesy of Ole Isacson, Harvard Medical School.]
Moreover, ApoE helped clear cholesterol from within cells, but how well it did so depended on the allele. ApoE binds to the outer cell surface, becomes lipidated, then carts the fats away. ApoE4 carries a lighter lipid load than E3 and E2, hence is worse at clearing intracellular lipids. In human astrocyte cultures, blocking NPC1 spurred cholesterol and triglyceride accumulations, and those co-localized with APP. Adding ApoE2 to the culture medium brought intracellular cholesterol and APP down to almost normal. Adding ApoE3 did so a little bit, while ApoE4 barely removed excess cholesterol and APP. “ApoE2 and ApoE3 can shuttle cholesterol out of the cell more effectively than ApoE4,” Isacson concluded. “This is very important for showing that functional differences in cholesterol lipid transport by ApoE isoforms correspond to the risk for developing LBD and AD.”
Kimberly Paul of the University of California, Los Angeles, took a different approach to learning about lipids in PD. She used mass spectrometry data of the metabolomes of serum samples from 642 people with early stage PD and 277 controls from UCLA’s Parkinson’s Environment and Genes study (Paul et al., 2022). Analyzing the data through a metabolome-wide association study, she found 104 of 4,762 identified metabolites to be specific to PD. Lo and behold, quite a few fell into lipid metabolism pathways.
Among the metabolites most associated with PD were various lysoPC species from the Lands cycle. People with PD had up to four times more of these lipids in their blood than did controls. LysoPCs also turned up at higher levels in the substantia nigra in a rat model of PD (Farmer et al., 2015). Functionally, these lipid metabolites are a “find me” signal on apoptotic cells to cue macrophages and microglia for phagocytosis. They have been linked to neuroinflammation and demyelination. "We are still teasing out the chain of events and mechanisms for lipid dyshomeostasis in PD, but lysoPCs seem to play a role,” Paul noted.
Could Choline Help Fix Lipid Dysfunction?
While phosphatidylcholine and lysoPC metabolism were disturbed in AD and PD, other signs point to perturbations in the lipid’s precursor molecule, choline. Carrying a mutation in the choline synthesis enzyme PEMT, and consuming insufficient choline in one's diet, are both linked to AD (Bi et al., 2011; Yuan et al., 2022). In amyloidosis mice, withholding dietary choline altered synaptic transmission and upped amyloid plaques and phospho-tau species, whereas APP/PS1 mice fed a high choline diet had fewer plaques and better spatial memory (Dave et al., 2023; Velazquez et al., 2019).
At AD/PD, Tsai reported similar results in APOE4 knock-in 5xFAD mice. A diet rich in choline cut amyloid load and lowered levels of the lipid droplet-associated protein perilipin1 in brain tissue. These findings mirror a reduction in lipid droplets Tsai saw within iPSC-derived human APOE4/4 astrocytes after adding choline to the culture medium (Mar 2021 news).
Taking choline supplements to treat or prevent AD has not been shown to work in clinical studies, though some are ongoing. It is unclear if dietary supplementation could overcome a deficit in choline transport or utilization, and, if so, how much choline would be required for a treatment benefit, McIntire wrote to Alzforum.—Chelsea Weidman Burke
References
News Citations
- Massive GWAS Meta-Analysis Digs Up Trove of Alzheimer’s Genes
- GWAS, GWAX: bioRχiv Hosts Bonanza of Alzheimer’s Genetics
- Lewy Body Dementia Shares Risk Genes with Alzheimer’s, Parkinson’s
- Droplets of Unsaturated Fats Burden Human ApoE4 Astrocytes
Mutations Citations
Research Models Citations
Paper Citations
- Bankaitis VA. The Cirque du Soleil of Golgi membrane dynamics. J Cell Biol. 2009 Jul 27;186(2):169-71. PubMed.
- Yassine HN, Croteau E, Rawat V, Hibbeln JR, Rapoport SI, Cunnane SC, Umhau JC. DHA brain uptake and APOE4 status: a PET study with [1-(11)C]-DHA. Alzheimers Res Ther. 2017 Mar 23;9(1):23. PubMed.
- Kaya I, Brinet D, Michno W, Başkurt M, Zetterberg H, Blenow K, Hanrieder J. Novel Trimodal MALDI Imaging Mass Spectrometry (IMS3) at 10 μm Reveals Spatial Lipid and Peptide Correlates Implicated in Aβ Plaque Pathology in Alzheimer's Disease. ACS Chem Neurosci. 2017 Dec 20;8(12):2778-2790. Epub 2017 Oct 4 PubMed.
- Kaya I, Brinet D, Michno W, Syvänen S, Sehlin D, Zetterberg H, Blennow K, Hanrieder J. Delineating Amyloid Plaque Associated Neuronal Sphingolipids in Transgenic Alzheimer's Disease Mice (tgArcSwe) Using MALDI Imaging Mass Spectrometry. ACS Chem Neurosci. 2017 Feb 15;8(2):347-355. Epub 2017 Jan 10 PubMed.
- Michno W, Wehrli PM, Zetterberg H, Blennow K, Hanrieder J. GM1 locates to mature amyloid structures implicating a prominent role for glycolipid-protein interactions in Alzheimer pathology. Biochim Biophys Acta Proteins Proteom. 2018 Sep 28; PubMed.
- Wehrli PM, Ge J, Michno W, Koutarapu S, Dreos A, Jha D, Zetterberg H, Blennow K, Hanrieder J. Correlative Chemical Imaging and Spatial Chemometrics Delineate Alzheimer Plaque Heterogeneity at High Spatial Resolution. JACS Au. 2023 Mar 27;3(3):762-774. Epub 2023 Mar 7 PubMed.
- Blanchard JW, Akay LA, Davila-Velderrain J, von Maydell D, Mathys H, Davidson SM, Effenberger A, Chen CY, Maner-Smith K, Hajjar I, Ortlund EA, Bula M, Agbas E, Ng A, Jiang X, Kahn M, Blanco-Duque C, Lavoie N, Liu L, Reyes R, Lin YT, Ko T, R'Bibo L, Ralvenius WT, Bennett DA, Cam HP, Kellis M, Tsai LH. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature. 2022 Nov;611(7937):769-779. Epub 2022 Nov 16 PubMed. Correction.
- Paul KC, Zhang K, Walker DI, Sinsheimer JS, Yu Y, Kusters C, DelRosario I, Folle AD, Keener AM, Bronstein J, Jones DP, Ritz B. Untargeted serum metabolomics reveals novel metabolite associations and disruptions in amino acid and lipid metabolism in Parkinson's disease. 2022 Dec 30 10.1101/2022.12.29.22284028 (version 1) medRxiv.
- Farmer K, Smith CA, Hayley S, Smith J. Major Alterations of Phosphatidylcholine and Lysophosphotidylcholine Lipids in the Substantia Nigra Using an Early Stage Model of Parkinson's Disease. Int J Mol Sci. 2015 Aug 12;16(8):18865-77. PubMed.
- Bi XH, Zhao HL, Zhang ZX, Zhang JW. PEMT G523A (V175M) Is Associated with Sporadic Alzheimer's Disease in a Chinese Population. J Mol Neurosci. 2011 Sep 1; PubMed.
- Yuan J, Liu X, Liu C, Ang AF, Massaro J, Devine SA, Auerbach SH, Blusztajn JK, Au R, Jacques PF. Is dietary choline intake related to dementia and Alzheimer's disease risk: results from the Framingham Heart Study. Am J Clin Nutr. 2022 Aug 2;116(5):1201-7. PubMed.
- Dave N, Judd JM, Decker A, Winslow W, Sarette P, Villarreal Espinosa O, Tallino S, Bartholomew SK, Bilal A, Sandler J, McDonough I, Winstone JK, Blackwood EA, Glembotski C, Karr T, Velazquez R. Dietary choline intake is necessary to prevent systems-wide organ pathology and reduce Alzheimer's disease hallmarks. Aging Cell. 2023 Feb;22(2):e13775. Epub 2023 Jan 15 PubMed.
- Velazquez R, Ferreira E, Knowles S, Fux C, Rodin A, Winslow W, Oddo S. Lifelong choline supplementation ameliorates Alzheimer's disease pathology and associated cognitive deficits by attenuating microglia activation. Aging Cell. 2019 Dec;18(6):e13037. Epub 2019 Sep 27 PubMed.
External Citations
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
Henan Academy of Innovations in Medical Science
It is great to see that more attention is paid to this important research area in AD. Shifts in lipid metabolism and biomarkers have been observed for a long time, but have not received much attention (Nasaruddin et al., 2018). What is odd in this article is that the obvious reason for this shift is not mentioned. Lipids are energy-rich molecules, and a shift of lipid metabolism clearly points toward issues with cell metabolism. We know that the AD brain reduces energy turnover and glucose uptake as shown in FDG-PET studies (Mosconi et al., 2010). Growth-factor signaling such as NGF or insulin in the brains of AD patients has been shown many times (Neth and Craft, 2017; Holscher, 2014), and nasal application of insulin already showed some improvements (Dhamoon et al., 2009). The growth-factor desensitization reduces glucose utilization, and cells start to switch to other forms of energy supply, such as amino acids (Maszka et al., 2023) and fatty acids (Nasaruddin et al., 2018).
A ketone diet improves cognition, as these energy-rich molecules can bypass the block of glucose uptake and supply neurons with the needed energy (Yin et al., 2016). A recent phase II clinical trial giving AD patients a diet rich in different types of energy-rich molecules that can bypass the block in glucose utilization has shown a 29 percent improvement in the ADAS-Cog after only 84 days (P = 0.00001) (Yulug et al., 2023), which beats the rather small improvement of lecanemab in 1,800 patients after 18 months of treatment. Instead of continuously staring at beta-amyloid as the solution to all problems, it is time to take a look at the physiological changes in the brains of AD. This will open up the developments of novel therapies that are non-toxic and work in most patients.
Instead of continuously staring at beta-amyloid as the solution to all problems, it is time to take a look at the physiological changes in the brains of AD. This will open up the developments of novel therapies that are non-toxic and work in most patients.
References:
Nasaruddin ML, Pan X, McGuinness B, Passmore P, Kehoe PG, Hölscher C, Graham SF, Green BD. Evidence That Parietal Lobe Fatty Acids May Be More Profoundly Affected in Moderate Alzheimer's Disease (AD) Pathology Than in Severe AD Pathology. Metabolites. 2018 Oct 26;8(4) PubMed.
Mosconi L, Berti V, Glodzik L, Pupi A, De Santi S, de Leon MJ. Pre-clinical detection of Alzheimer's disease using FDG-PET, with or without amyloid imaging. J Alzheimers Dis. 2010;20(3):843-54. PubMed.
Neth BJ, Craft S. Insulin Resistance and Alzheimer's Disease: Bioenergetic Linkages. Front Aging Neurosci. 2017;9:345. Epub 2017 Oct 31 PubMed.
Hölscher C. Insulin, incretins and other growth factors as potential novel treatments for Alzheimer's and Parkinson's diseases. Biochem Soc Trans. 2014 Apr;42(2):593-9. PubMed.
Dhamoon MS, Noble JM, Craft S. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology. 2009 Jan 20;72(3):292-3; author reply 293-4. PubMed.
Maszka P, Kwasniak-Butowska M, Cysewski D, Slawek J, Smolenski RT, Tomczyk M. Metabolomic Footprint of Disrupted Energetics and Amino Acid Metabolism in Neurodegenerative Diseases: Perspectives for Early Diagnosis and Monitoring of Therapy. Metabolites. 2023 Mar 1;13(3) PubMed.
Yin JX, Maalouf M, Han P, Zhao M, Gao M, Dharshaun T, Ryan C, Whitelegge J, Wu J, Eisenberg D, Reiman EM, Schweizer FE, Shi J. Ketones block amyloid entry and improve cognition in an Alzheimer's model. Neurobiol Aging. 2016 Mar;39:25-37. Epub 2015 Dec 7 PubMed.
Yulug B, Altay O, Li X, Hanoglu L, Cankaya S, Lam S, Velioglu HA, Yang H, Coskun E, Idil E, Nogaylar R, Ozsimsek A, Bayram C, Bolat I, Oner S, Tozlu OO, Arslan ME, Hacimuftuoglu A, Yildirim S, Arif M, Shoaie S, Zhang C, Nielsen J, Turkez H, Borén J, Uhlén M, Mardinoglu A. Combined metabolic activators improve cognitive functions in Alzheimer's disease patients: a randomised, double-blinded, placebo-controlled phase-II trial. Transl Neurodegener. 2023 Jan 26;12(1):4. PubMed.
Make a Comment
To make a comment you must login or register.