Cohort LEADS Toward Better Understanding of Sporadic Early Onset AD
Quick Links
When people think of early onset Alzheimer’s disease (EOAD), autosomal-dominant mutations in the APP or presenilin genes come to mind. But these account for fewer than 15 percent of EOAD cases. For the remainder, the Longitudinal Early-Onset Alzheimer’s Disease Study (LEADS), a collaboration led by Liana Apostolova at Indiana University School of Medicine, Indianapolis, Brad Dickerson of Massachusetts General Hospital, Boston, Gil Rabinovici at the University of California, San Francisco, and Maria Carrillo of the Alzheimer’s Association aims to characterize their disease and decipher its root cause. At AAIC, held July 16-20, scientists shared baseline and longitudinal data on genetics, imaging, and fluid biomarkers.
- LEADS aims to study 400 adults, ages 40 to 64, who have Alzheimer’s but no autosomal-dominant AD mutation.
- Some carry FTD or Parkinson's genes.
- Many likely became amyloid-positive in their 30s.
- The earlier EOAD begins, the faster it gets worse.
“Our ultimate goal is to launch clinical trials," Apostolova wrote to Alzforum. "We consider EOAD to be the ideal Phase 2 cohort, because these young patients have a ‘pure’ form of AD and few medical comorbidities, yet advance rapidly” (full comment below).
What is Early Onset AD?
Sporadic EOAD strikes before age 65. Because of their young age, people with EOAD have largely been excluded from research. They might make for a suitable clinical trial population, because they have few co-pathologies and their disease progresses rapidly. What causes EOAD is unknown, but scientists believe the answers may lie in undiscovered genetic risk variants.
While most adults with EOAD primarily have memory problems, a greater proportion than is the case in LOAD are diagnosed with rare, non-amnestic subtypes, including primary progressive aphasia (PPA) or posterior cortical atrophy (PCA; Jul 2012 conference news). At AAIC, Angelina Polsinelli of Indiana U described two such cases from LEADS.
A 55-year-old man developed progressive memory and language problems over 18 months, which cost him his job because he mispronounced words, asked people to repeat themselves, and could no longer follow directions. His Mini-Mental State Exam (MMSE) score was 17 out of 30, working memory and language being weakest. He scored 29 out of 60 on the Boston Naming Test, on which controls his age score in the 50s. MRI showed an abnormally small left lateral-temporal lobe, an area that is key to speech and language and is often atrophied in PPA. Since his cerebrospinal fluid Aβ42/40 ratio was low, CSF total tau and phospho-tau181 high, and amyloid and tau PET scans positive, his diagnosis was PPA due to AD.
A 56-year-old woman struggled with depth perception and finding her way. She'd swerve out of her lane while driving and got lost going to the grocery store or doctor’s appointments. Her memory began declining two years later. Her MMSE was 22, with visuospatial perception faring worst. When copying an image, her version appeared “exploded,” a quintessential sign of PCA (McMonagle and Kertesz, 2006). Indeed, MRI showed shrunken temporal, parietal, and occipital cortices. Her low CSF Aβ42/40 ratio, high CSF t-tau and ptau-181, and positive amyloid and tau PET scans prompted a diagnosis of PCA due to AD.

Looks Like PCA. A woman with EOAD rendered a typically disjointed copy of a drawing and was diagnosed with PCA. [Courtesy of Angelina Polsinelli, Indiana University.]
The LEADS Cohort
Most EOAD cases are not so easily defined. PPA and PCA each account for 6 percent of the LEADS cohort, which was created to characterize EOAD cases that lack a clear clinical diagnosis. Designed after the Alzheimer’s Disease Neuroimaging Initiative (ADNI), LEADS will enroll 400 and 100 cognitively normal adults, ages 40 to 64, at 18 U.S. sites. Participants must not carry known mutations for autosomal-dominant AD or frontotemporal dementia. Researchers will follow controls for two years, impaired participants for four, collecting PET and MRI data, cerebrospinal fluid and blood, and administering cognitive and neuropsychologic tests.
LEADS began enrolling in 2018 and expected slow enrollment over three years because EOAD is rare, and COVID slowed things down further, Apostolova explained. Complicating the issue, 25 percent of those who were recruited tested negative for amyloid. These early onset non-AD volunteers came as a surprise to the LEADS investigators. They decided to call them EOnonAD and keep them in the study.
At AAIC, Dustin Hammers of Indiana U presented the baseline characteristics of the cohort (Hammers et al., 2023). As of June 2022, researchers had enrolled 89 controls, 212 people with clinically diagnosed EOAD, and 70 with EOnonAD. Participants were in their mid- to late-50s on average, half were women, 86 percent Caucasian.
At baseline, the EOAD group had worse memory scores than the EOnonAD group, averaging 22 on the MMSE versus 25.5 for the latter. Specifically, they did worse on episodic memory, executive function, and attention. These differences held true after accounting for the severity of global cognitive decline, age, sex, and years of education.
APOE genotype seemed to play a big role. Compared to EOnonAD participants, five times as many people with EOAD were APOE4 homozygous. Half as many had an E2 allele. In the EOAD group, 54 percent carried an APOE4 allele. In EOnonAD and controls, 41 percent did. “We were surprised by the high level of E4 heterozygosity in these two groups,” said Hammers.
What is EOnonAD? The LEAD investigators suspect an FTD-like disease, because these participants had more neuropsychiatric symptoms than their EOAD counterparts. In Amsterdam, Polsinelli reported that they were more apathetic and irritable, had less impulse control, and took more mood stabilizers at baseline (Polsinelli et al., 2023). “Although affective symptoms were common in all early onset AD cases, they were more so in EOnonAD,” she said.
In the EOnonAD group, 62 were not diagnosed with PPA, PCA, or non-amnestic dementia. Some of them had cognitive, fluid marker, and imaging characteristics of FTD. Hee Jin Kim, a visiting professor at Indiana U from South Korea’s Sungkyunkwan University, reported that some scored poorly on executive function, visuospatial, and speed/attention tasks, had high CSF and plasma levels of the neurodegeneration marker neurofilament light, and marked atrophy in their frontoparietal lobes.
What Does Genetics Say?
Genotyping, too, suggested an FTD component to EOnonAD. Kelly Nudelman of Indiana U used whole-exome sequencing in search of known pathogenic mutations. In the EOnonAD sample, two carried pathogenic variants in progranulin (GRN), one in MAPT, and two had expansions in C9ORF72. The same search in the EOAD group netted three participants carrying pathogenic PSEN1 variants.
Genetic screening was not an enrollment criterion in LEADS, hence these eight people's genotype status was determined after they had had baseline data collected. They were subsequently excluded from further data analysis and referred to the ALLFTD Study or the Dominantly Inherited Alzheimer Network. No other FTD or ADAD mutation carriers have turned up since, according to Apostolova.
Some other genetic variants already appear to be involved. One person with EOAD and one with EOnonAD carried a variant in the sphingolipid hydrolase gene SMPD1; variants in this gene cause Niemann-Pick’s disease in homozygous carriers. Fifteen people with EOAD and two with EOnonAD carried a dominant pathogenic mutation in the Parkinson’s risk genes GBA or LRRK2. Another six, four EOAD and two EOnonAD, had a recessive variant in a copy of one of three other PD genes—PRKN, PARK7, or PRKRA.
“In a small subset of LEADS cases, the SMPD1 and PD variants may contribute to overall risk for early onset AD, without necessarily being the sole cause,” Nudelman concluded. Strangely, at baseline, only two PD variant carriers had motor symptoms—tremor or slow gait—though nine had peripheral neuropathy, REM-sleep behavior disorder, or other non-motor signs of PD.
Still, most EOAD and EOnonAD cases could not be explained by known pathogenic variants in genes related to neurodegenerative diseases. Two AD risk variants in TREM2, R47H and R62H, turned up in the cohort, but were as prevalent in controls as in symptomatic participants. “We think EOAD is enriched with novel genetic risk factors that might inform our understanding of disease mechanisms and identify novel pathways and drug targets,” Rabinovici wrote to Alzforum (comment below).
Clues from Imaging
Volumetric brain scans revealed differences between EOAD and EOnonAD. Alexandra Touroutoglou, MGH, spotted a pattern whereby the posterior cingulate cortex, lateral temporal cortex, posterior temporal lobe, inferior parietal lobe, and precuneus were smaller in EOAD than in controls. In contrast, EOnonAD participants had no signs of cortical degeneration. This atrophy signature distinguished EOAD from controls and from EOnonAD more accurately than did scanning for the typical atrophy pattern seen in people with LOAD (Dickerson et al., 2009). “The LOAD signature captured atrophy less well than our new EOAD signature. This supports the use of a measure tailored to EOAD, especially to detect small longitudinal changes, as might be expected from disease-modifying therapies,” Touroutoglou said (image below).

EOAD Signature. Nine cortical regions withered in people with EOAD (top), but not in EOnonAD (bottom). [Courtesy of Alexandra Touroutoglou, Massachusetts General Hospital.]
Greater atrophy of the EOAD signature regions correlated with poorer scores on the Clinical Dementia Rating–Sum of Boxes (CDR-SB) and MMSE. Touroutoglou will follow this cohort and look for different signatures in subtypes, such as PCA and PPA.
Comparing amyloid plaque and neurofibrillary tangle pathology between EOAD and EOnonAD, UCSF’s Renaud La Joie saw that, on baseline PET, all EOnonAD participants were amyloid-negative and most were tau-negative. In contrast, everyone with EOAD was amyloid-positive, and 90 percent were tau-positive (see image below). Plaque and tangle load varied greatly in EOAD, but more amyloid generally tracked with more tangles.

Pretty Separate. EOnonAD participants (orange) were amyloid-negative (x-axis) and most were tau-negative (y-axis). Most people with EOAD (black) were amyloid-positive, and varied greatly in their tangle loads. [Courtesy of Renaud La Joie, University of California, San Francisco.]
What might influence tangle load? Age at onset showed a dramatic link. Compared to people who became symptomatic in their mid-60s, people for whom this happened in their late 40s or early 50s had more cortical tangles at the baseline point of the LEADS study, and for 17 who had a follow-up scan 3.5 years later, they accumulated additional tangles faster if their age at onset was earlier (see image below). Daniel Schonhaut, also at UCSF, predicted that a 55-year-old person with EOAD would amass tangles twice as fast as someone a decade older when all other demographics matched, even if they had a similar tau load on their first scan.
Why? Perhaps tau aggregates more rapidly in younger brains. Previously, scientists correlated seeding activity of tau in brain extracts with AAO, with seeds from younger people being more aggressive (Jun 2020 news). Rabinovici thinks stronger network connectivity in younger people might also play a role. “If tau spreads trans-synaptically, the higher levels of neocortical activity in young patients may provide many more neuronal pathways for tau to spread,” he hypothesized.
Hammers found that 45- to 50-year-olds with EOAD had worse memory, visuospatial function, and attention than their older counterparts, suggesting that earlier onset signals more severe disease. This phenomenon matches prior findings in LOAD. People who developed AD between ages 65 to 74 dropped 2.0 points annually on the MMSE, while those who first showed symptoms over age 75 slipped 1.4 points per year (Stanley et al., 2019). “Age of onset should be considered a continuous variable, as the association between earlier onset and worse disease progression doesn’t start or stop at age 65,” La Joie said.
Age at onset did not influence baseline amyloid burden or amyloid accumulation over time.
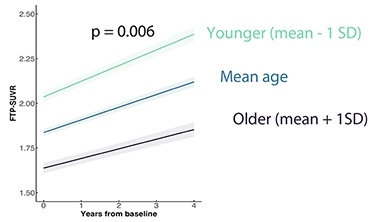
Getting Worse Faster. People whose EOAD began in their late 40s to early 50s (green) had higher tangle load at the LEADS baseline scan and accumulate tau faster than those whose disease began in their early 60s (navy). [Courtesy of Renaud La Joie, University of California, San Francisco.]
Digging deeper into how tau aggregates in EOAD, Schonhaut found that, at baseline, tangles were primarily in the basal-lateral-temporal lobe, parietal association cortex, and mid-frontal gyrus. Over an average of three years, every region except the amygdala accumulated tangles, with the frontal, temporal, lateral, and occipital lobes amassing them quickest. Regions in the first three were part of the EOAD atrophy signature Touroutoglou identified.
Wondering how LEADS baseline characteristics other than age related to tau pathology, Schonhaut looked at sex, amyloid burden, CDR-SB score, and APOE genotype. Only the latter stood out. Over their follow-up, homozygous APOE4 carriers accumulated tangles twice as fast as noncarriers, even after accounting for their baseline amyloid PET load. Scientists are just beginning to appreciate how ApoE4 worsens tau pathology through Aβ-dependent and -independent pathways (Feb 2023 news; Jul 2022 news; Yamazaki et al., 2019).
As for amyloid accumulation, it is strikingly similar in EOAD and LOAD. When UCSF’s Ganna Blazhenets looked at this question, she did not notice this at first, because people with EOAD had more baseline amyloid yet added plaques more slowly than their LOAD counterparts in ADNI. However, Blazhenets realized that EOAD participants were, on average, farther up their amyloid staging curve than ADNI participants.
“Patients with EOAD face a longer path to diagnosis,” Rabinovici said. “Clinicians often don’t have AD on their radar because of the patient’s young age and atypical presentation, often with executive functioning, language, or visuospatial problems rather than memory issues.” Younger people also have high brain reserves and few co-pathologies, enabling them to tolerate a high amyloid burden before showing symptoms, he added. In fact, Blazhenets found that EOAD and LOAD participants with similar baseline amyloid loads accumulated plaques at the same rate (see image below).

Cut From Same Cloth? Amyloid accumulation tends to be detected at different time points in early and late-onset AD (top), since EOAD is often diagnosed at a later stage. When compared at the same baseline amyloid load (bottom left), EOAD and LOAD had the same accumulation rates in a linear mixed-effects model (bottom right). [Courtesy of Ganna Blazhenets, University of California, San Francisco.]
Given that, Blazhenets calculated that the average 75-year-old person in ADNI became amyloid-positive at age 52, while the average 59-year-old in LEADS had turned positive at age 32.
Being amyloid- and tau-negative and unlikely to carry APOE4, people with EOnonAD resemble another mysterious group with suspected non-Alzheimer's pathophysiology. Remember SNAP (Sep 2015 news)? These people have biomarkers of neurodegeneration, yet can be cognitively normal, whereas EOnonAD means dementia. “Technically, some of the EOnonAD individuals in LEADS would likely fall in the SNAP category,” Polsinelli said.
Fluid Biomarkers
If amyloid deposition in EOAD parallels that of LOAD, how about CSF and blood markers? At baseline, they do, too, according to Jeff Dage at Indiana U. Compared to controls, EOAD participants not only had low CSF Aβ42/40 ratios and high CSF p-tau181, t-tau, and NfL, but also abnormal reads of other markers of neuronal health, such as presynaptic protein SNAP25, the postsynaptic protein neurogranin, and the neuronal calcium sensor Vilip1. Only NfL consistently correlated with worse cognition as per CDR-SB, ADAS-Cog1, or MoCa (Dage et al., 2023).
In EOnonAD participants, CSF NfL was slightly up over controls, indicating some neurodegeneration. All other markers were normal, in keeping with a non-AD etiology.
Plasma biomarkers told the same tale. Paige Logan, in Apostolova’s lab, reported that EOAD baseline p-tau231, NfL, and GFAP were high and correlated with more tangles, but not plaques, while an uptick in NfL came with cortical gray-matter thinning. Ralitsa Kostadinova in the same lab found that, in EOAD, these three markers tracked with worse scores on the ADAS-Cog13, MoCA, MMSE, and CDR-SB.
In EOnonAD, only plasma NfL and GFAP were up, and correlated with cognitive decline.
Notably, marker levels differed by sex. Sára Nemes in the Apostolova lab detected more NfL and GFAP in the blood, and more p-tau181, t-tau, neurogranin, and Vilip1 in the CSF of women with EOAD than in blood and CSF from men. Even women with EOnonAD had higher levels of plasma GFAP than men, suggesting that these sex differences occur regardless of amyloid positivity. Nemes thinks this reflects worse pathology or neurodegeneration in women.
“This study provides the first comprehensive analysis of fluid biomarkers in sporadic EOAD. It can inform clinical trial designs that incorporate such markers in patient selection or as endpoints,” Dage said.
LEADing the Way
Apostolova said that as of July 2023, LEADS had enrolled 99 out of 100 cognitively normal controls, 361 people with EOAD, and 112 with EOnonAD. Hammers expects the study to finish enrolling by this fall once it has included 400 EOAD cases. In May 2023, five research papers and a commentary on data generated by LEADS appeared together in a special issue of Alzheimer’s & Dementia. It will contain 11 manuscripts total.
LEADS is set to add five international study sites: University College London, Vrije University Medical Center in Amsterdam, Lund University in Sweden, Sant Pau Memory Center in Barcelona, and Fleni, a nonprofit neurological hospital in Buenos Aires. This is to establish a global network for future cohort studies and, ultimately, clinical trials for EOAD. “The first round of LEADS is observational to optimize the planning of trial outcomes, with the goal of renewing the study to add trial units, like DIAN-TU,” Rabinovici wrote.—Chelsea Weidman Burke
References
News Citations
- Researchers Join to Draw Posterior Cortical Atrophy Out of Shadows
- Tau: Why Alzheimer’s Worsens Fast in Some, Slowly in Others
- Secreted by Neurons, ApoE4 Makes Tangles and Degeneration Worse
- Does Tangle Progression Follow ApoE Expression?
- Suspected Non-Alzheimer Pathophysiology: It’s Not Exactly a Snap
Mutations Citations
Paper Citations
- McMonagle P, Kertesz A. Exploded drawing in posterior cortical atrophy. Neurology. 2006 Nov 28;67(10):1866. PubMed.
- Hammers DB, Eloyan A, Taurone A, Thangarajah M, Beckett L, Gao S, Kirby K, Aisen P, Dage JL, Foroud T, Griffin P, Grinberg LT, Jack CR Jr, Kramer J, Koeppe R, Kukull WA, Mundada NS, La Joie R, Soleimani-Meigooni DN, Iaccarino L, Murray ME, Nudelman K, Polsinelli AJ, Rumbaugh M, Toga A, Touroutoglou A, Vemuri P, Atri A, Day GS, Duara R, Graff-Radford NR, Honig LS, Jones DT, Masdeu J, Mendez MF, Womack K, Musiek E, Onyike CU, Riddle M, Rogalski E, Salloway S, Sha SJ, Turner RS, Wingo TS, Wolk DA, Carrillo MC, Dickerson BC, Rabinovici GD, Apostolova LG, LEADS Consortium. Profiling baseline performance on the Longitudinal Early-Onset Alzheimer's Disease Study (LEADS) cohort near the midpoint of data collection. Alzheimers Dement. 2023 May 31; PubMed.
- Polsinelli AJ, Wonderlin RJ, Hammers DB, Garcia AP, Eloyan A, Taurone A, Thangarajah M, Beckett L, Gao S, Wang S, Kirby K, Logan PE, Aisen P, Dage JL, Foroud T, Griffin P, Iaccarino L, Kramer JH, Koeppe R, Kukull WA, La Joie R, Mundada NS, Murray ME, Nudelman K, Soleimani-Meigooni DN, Rumbaugh M, Toga AW, Touroutoglou A, Vemuri P, Atri A, Day GS, Duara R, Graff-Radford NR, Honig LS, Jones DT, Masdeu J, Mendez MF, Womack K, Musiek E, Onyike CU, Riddle M, Rogalski E, Salloway S, Sha SJ, Turner RS, Wingo TS, Wolk DA, Carrillo MC, Dickerson BC, Rabinovici GD, Apostolova LG, LEADS Consortium. Baseline neuropsychiatric symptoms and psychotropic medication use midway through data collection of the Longitudinal Early-Onset Alzheimer's Disease Study (LEADS) cohort. Alzheimers Dement. 2023 Nov;19 Suppl 9:S42-S48. Epub 2023 Jun 9 PubMed.
- Dickerson BC, Bakkour A, Salat DH, Feczko E, Pacheco J, Greve DN, Grodstein F, Wright CI, Blacker D, Rosas HD, Sperling RA, Atri A, Growdon JH, Hyman BT, Morris JC, Fischl B, Buckner RL. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex. 2009 Mar;19(3):497-510. PubMed.
- Stanley K, Whitfield T, Kuchenbaecker K, Sanders O, Stevens T, Walker Z. Rate of Cognitive Decline in Alzheimer's Disease Stratified by Age. J Alzheimers Dis. 2019;69(4):1153-1160. PubMed.
- Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019 Sep;15(9):501-518. Epub 2019 Jul 31 PubMed.
- Dage JL, Eloyan A, Thangarajah M, Hammers DB, Fagan AM, Gray JD, Schindler SE, Snoddy C, Nudelman KN, Faber KM, Foroud T, Aisen P, Griffin P, Grinberg LT, Iaccarino L, Kirby K, Kramer J, Koeppe R, Kukull WA, La Joie R, Mundada NS, Murray ME, Rumbaugh M, Soleimani-Meigooni DN, Toga AW, Touroutoglou A, Vemuri P, Atri A, Beckett LA, Day GS, Graff-Radford NR, Duara R, Honig LS, Jones DT, Masdeu JC, Mendez MF, Musiek E, Onyike CU, Riddle M, Rogalski E, Salloway S, Sha SJ, Turner RS, Wingo TS, Wolk DA, Womack KB, Carrillo MC, Dickerson BC, Rabinovici GD, Apostolova LG, LEADS Consortium. Cerebrospinal fluid biomarkers in the Longitudinal Early-onset Alzheimer's Disease Study. Alzheimers Dement. 2023 Nov;19 Suppl 9:S115-S125. Epub 2023 Jul 25 PubMed.
External Citations
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
UCSF
The impetus for LEADS is twofold. First, we think sporadic early onset Alzheimer’s disease (EOAD) is an ideal population for AD trials and drug development. They have fewer co-pathologies, so AD is driving clinical decline. Biomarkers and clinical outcomes progress more quickly, so you can determine drug effects with smaller samples and shorter trial duration. Patients are generally highly motivated to participate in research. The first round of LEADS is observational to optimize the planning of trial outcomes, but the goal with renewal is to add a trials unit, à la DIAN-TU.
Second, we think EOAD is enriched with novel genetic risk factors that might inform our understanding of disease mechanisms and identify novel pathways and drug targets. As you saw, heritability is high, yet autosomal-dominant mutations are very rare, and some of our patients even have an APOE E2 genotype. These patients are getting AD without the two major risk factors (age and APOE4) and this is something we need to better understand. We plan to pursue whole-genome sequencing and additional cutting-edge genetic/genomic approaches to study this population.…More
An unexpected finding is that 25 percent of patients who meet clinical criteria for EOAD are amyloid-negative. Better understanding the causes of impairment in these individuals should inform clinical practice.
The addition of international sites is a new development that is very exciting and will advance all these goals.
Make a Comment
To make a comment you must login or register.