Microglia Control Synapse Number in Multiple Disease States
Quick Links
What causes synapses to wither in neurodegenerative diseases such as Alzheimer’s? Some researchers blame reactivation of microglia, which prune unneeded synapses during brain development. At the Society for Neuroscience meeting held in Chicago October 17-21, Beth Stevens of Boston Children’s Hospital, a pioneer in this field, presented new evidence to support the idea that these immune cells of the brain devour synapses in Alzheimer’s and Huntington’s diseases. Other researchers reported that excessive synapse consumption occurs in obesity as well, and that problems with developmental pruning may predispose children to disorders such as schizophrenia and autism. Several talks and posters described the mechanisms by which microglia control the number of synapses, including the identification of a protective “don’t eat me” signal. The topic generated buzz among attendees and appears to be gaining traction among neuroscientists, as evidenced by multiple groups now reporting similar data.
“This research has changed our understanding of microglia and their potential roles in neurodegenerative and neurodevelopmental disorders … the field is moving forward very fast and the new developments are exciting,” Jonathan Kipnis of the University of Virginia, Charlottesville, wrote to Alzforum. He studies interactions between immune cells and neurons. Ben Barres of Stanford University, California, agreed, noting, “The work not only provides a detailed mechanistic pathway for understanding how synapses are lost, but also one that is therapeutically targetable.”

Immune Molecules Dapple Brain.
Three different MHCI molecules (red, blue, and green) reveal distinct expression patterns in the brains of 6-week old mice. [Image courtesy of Carla Shatz and Science/AAAS.]
The idea that the immune system regulates synapse number dates back only about a decade. At that time, Carla Shatz of Stanford reported excessive synaptic plasticity in mice that lacked the major histocompatibility class I (MHCI) protein or its receptor PirB. Meanwhile Stevens, then working with Barres, described a synaptic bounty in young mice lacking the C1q or C3 proteins of the immune complement system, which signal microglia to snip synapses (see Nov 2007 conference news). Other groups have since confirmed that microglia touch, regulate, and engulf synapses (see Nov 2010 news; Dec 2013 news). Astrocytes get into the act as well, chewing up synapses during development using a different suite of receptors (see Dec 2013 conference news).
Some studies hinted that synapse ingestion might ramp back up in Alzheimer’s disease. Shatz reported synapse retention in AD model mice lacking PirB (see Sep 2013 news). Likewise, Cynthia Lemere at Brigham and Women’s Hospital, Boston, in collaboration with Stevens, found more synapses and better memory in APPPS1 mice lacking C3 (see Aug 2013 conference news). Recently, Stevens’ group tied elevated C1q to synaptic disconnection in the J20 mouse model (see Mar 2015 conference news).
At SfN, Stevens expanded on these data. In a Presidential Special Lecture before a packed hall, she noted that synapses in AD model mice bristle with excess C1q even at young ages, before amyloid plaques form. The fact that C3 knockouts are protected from the synaptic carnage normally seen in AD mice suggests that complement proteins mediate elimination, she said. To find out if Aβ plays a role, the researchers injected oligomers of the peptide into the ventricles of wild-type mice. Within three days, they saw a dramatic rise in C1q at synapses in stained brain sections from hippocampus, as well as a steep drop in synapse number. Microglia likely gobbled up these structures, as the researchers saw fluorescently labeled synapses inside these immune cells after Aβ treatment. Again, complement appeared to mediate loss, as mice missing the C1qA subunit maintained their synapses just fine after Aβ injections. Likewise, blocking C1q by passive immunotherapy before injecting Aβ protected synapses from destruction in live mice. Stevens did not say if the immunotherapy alone had any effect. In hippocampal slices, pre-treatment with the antibody preserved long-term potentiation. Altogether, the data implicate microglia and complement as the culprits in the disappearance of synapses in AD models, Stevens said. She added that it is still unknown what causes C1q to rise in these models, or how Aβ and C1q work together.
What about other neurodegenerative diseases? In a separate talk, Daniel Wilton in Stevens’ group extended the findings to mouse models of Huntington’s disease. In this condition, caused by a polyglutamine expansion in the huntingtin protein, people develop both movement and mental problems and striatal degeneration with age. Wilton used the Q175 HD mouse, which loses synapses but not neurons, and probably models early disease. By three months of age, C1q accumulated at synapses, and microglial activation ran wild in the striatum and motor cortex, he found. Microglia in these mice contained more synaptic material than did those in control animals, suggesting they were gorging on these connections. Crossing Q175 mice with CR3 knockouts protected synapses, as did injecting a C1q antibody every other day for a month. In future work, Wilton will examine whether protecting synapses slows the progression of the disease and helps preserve motor functions. When asked if the same mechanism occurs in human disease, Wilton said preliminary data reveal more complement at synapses in postmortem brain slices from HD patients than from healthy controls. He is also measuring complement levels in cerebrospinal fluid samples from patients to see if these proteins spike before synapses start to disappear.
Microglia may even thin out synapses in other health conditions. Obesity, a risk factor for cognitive decline and dementia, boosts inflammation throughout the body (see Sep 2015 news; AlzRisk analysis). Alexis Stranahan of the Medical College of Georgia, Augusta, wondered if that inflammation might activate microglia. To test this, she compared mice that ate chow containing 60 percent fat to those on a 10 percent fat diet for 12 weeks. The tubby mice had more C1q at synapses, and more microglia crowded those synapses, she found. In culture, microglia from the obese mice were more mobile and devoured more synapses, suggesting they had been activated for phagocytosis (see Hao et al., 2015). Inhibiting the C3 complement receptor prevented the synaptic stripping, Stranahan said. C3 is activated by C1q.
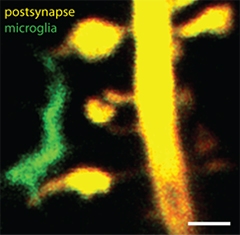
Intimate Contact.
Microglial processes (green) touch dendritic spines (yellow) in cultured hippocampal slices. Scale bar, 1 μm. [Image courtesy of Thomas Pfeiffer and Valentin Naegerl, University of Bordeaux.]
In addition to these late-life disorders, perturbations in synapse maintenance may also contribute to neurodevelopmental conditions such as schizophrenia and autism—though whether the problem is too few synapses or too many remains unclear. At SfN, Cornelius Gross of the European Molecular Biology Laboratory, Monterotondo, Italy, made a case for the latter. He reported that mice lacking the microglial fractalkine receptor, CX3CR1, maintained a greater number of feeble synapses. In these mice, brain activity coordinated poorly. The animals repeated actions over and over, and interacted poorly with littermates, traits reminiscent of autism (see Zhan et al., 2014). Why might this be? Fractalkine is a neuronal transmembrane protein that attracts microglia and is highly expressed during synaptogenesis. The CX3CR1 knockouts have fewer microglia in their brains, which may lead to deficient synaptic pruning, Gross said. Fractalkine expression in apoptotic neurons has previously been found to stimulate microglial phagocytosis (see Sokolowski et al., 2014). Gross noted that the failure to eliminate some synapses seems to prevent the strengthening of others, as though there is some limiting resource. This leads to overall weak synaptic connectivity, a pattern also seen in autism and other neurodevelopmental disorders. Together, the results hint that insufficient synaptic pruning by microglia could contribute to neurodevelopmental problems.
Other evidence implicates excessive loss of synapses. Stevens described a genetic study of 34,000 people with schizophrenia by Steve McCarroll of Harvard Medical School. It correlated polymorphisms near the C4 complement gene with disease risk. In particular, McCarroll saw a connection with the allele for the C4A subunit. This gene has undergone extensive duplication and deletion through the years, such that the number of copies vary widely from one person to another. He found that people lacking the allele for the C4A subunit had a low risk of the disease, whereas those with multiple copies had high risk. C4 participates in synaptic pruning just as C1q and C3 do, Stevens noted. The data suggest overzealous synapse elimination might underlie schizophrenia.
Similarly, Kim McAllister at UC Davis pointed a finger at excessive loss of synapses in neurodevelopmental disorders. Myka Estes in McAllister’s lab studies a mouse model of maternal immune activation (MIA), in which researchers inject a viral mimic into pregnant females to stimulate inflammation (see Malkova et al., 2012). Some researchers believe maternal inflammation sets the stage for later schizophrenia in children, and indeed, the adult offspring of these mice exhibit behaviors reminiscent of autism and schizophrenia. In 2013, McAllister and colleagues found that the offspring also had more major histocompatibility class I (MHCI) marking synapses, and only half as many synapses as controls, McAllister reported (see Elmer et al., 2013).
In Chicago, McAllister said that the researchers had linked these changes to a doubling of the cytokine IL-1β in the offspring. In support of a role for IL-1β, treating cultured neurons with this cytokine boosted MHCI and reduced the number of synapses on the cell surface. Conversely, lowering MHCI preserved synapses from the harmful effects of IL-1β. McAllister hypothesized that maternal infection pumps up IL-1β levels, leading to elevated MHCI and a corresponding loss of synapses. IL-1β is also elevated in the blood and brain of people with schizophrenia and autism, she noted.
Data from other groups supports a role for IL-1β in synaptic pruning. In a poster, Nasr Farooqi from the lab of Edward Ruthazer at McGill University reported that the cytokine prevented synapse stabilization in zebrafish larva. Stimulating inflammation in these animals caused microglia to activate and pump out IL-1β. Axons initially became more mobile and put out more feelers, but in succeeding days their axonal arbors thinned out, suggesting that new synapses failed to stabilize, said Ruthazer. Knockdown of IL-1β or deletion of microglia prevented this loss. Intriguingly, IL-1β has also been implicated in loss of synapses in obese mice (see Erion et al., 2014).
Can synapses protect themselves from overeager microglial pruning? It appears so. Emily Lehrman in Stevens’ group described a protective signal. In the periphery, transmembrane CD47 acts as a “don’t-eat-me” signal for many cell types, binding to signal regulatory protein α (SIRPα) on macrophages to inhibit engulfment. Lehrman wondered if CD47 could play a similar role in the brain. Consistent with this, she found high CD47 at some but not all synapses in the visual system of mice five days after birth, during the peak of synaptic pruning, as well as high SIRPα in microglia. By two months of age, both CD47 knockouts and SIRPα knockouts had lost more synapses than wild-type mice, fitting with the idea that these are protective molecules, she reported. In addition, blocking SIRPα boosted microglial appetites for synapses in vitro. In future work, Lehrman will assess whether these changes in synapse number have consequences for behavior. Other studies have found that CD47 knockouts are less sociable, she noted (see Koshimizu et al., 2014).
Overall, the emerging picture suggests that microglia affect synapses in both development and disease states. Could they regulate synaptic plasticity in healthy adults as well? Thomas Pfeiffer, working in Valentin Nägerl’s team at the University of Bordeaux, France, presented some mouse evidence that they might. Using two-photon imaging in hippocampal slices from healthy adults, he found that after stimulating long-term potentiation of synapses, microglia scanned their environment more actively, putting out more processes. Surprisingly, the microglia made fewer contacts with synapses, but maintained each contact for longer. Pfeiffer could prevent these microglial changes by blocking NMDA receptors, which mediate synaptic remodeling, demonstrating that the immune cells were responding to LTP. Next, he will use super-resolution microscopy to investigate whether synapses touched by microglia share some common feature, and what happens to them after this contact, he told Alzforum.
Researchers agree that they have only begun to scratch the surface of how microglia and astrocytes may affect synapses. “The field is in its infancy,” Kipnis wrote to Alzforum. “It may be too early to understand exactly what we are seeing, but it’s something we were previously blind to.”—Madolyn Bowman Rogers
References
News Citations
- San Diego: MHC Class I and Complement—Holding Down Second Jobs in the Synapse
- No Rest for Microglia: These Immune Cells Manage Healthy Synapses
- Beyond Neighborhood Watch—Microglia Nurture Synapses
- Glial Cells Refine Neural Circuits
- Immune Receptor Binds Aβ Oligomers, Spurs Synaptic Loss
- Curbing Innate Immunity Boosts Synapses, Cognition
- Microglia Rely on Mixed Messages to Select Synapses for Destruction
- Extra Weight in Midlife Hastens Onset of Alzheimer’s Disease
Research Models Citations
Paper Citations
- Hao S, Dey A, Yu X, Stranahan AM. Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain Behav Immun. 2015 Aug 31; PubMed.
- Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, Vyssotski AL, Bifone A, Gozzi A, Ragozzino D, Gross CT. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. 2014 Mar;17(3):400-6. Epub 2014 Feb 2 PubMed.
- Sokolowski JD, Chabanon-Hicks CN, Han CZ, Heffron DS, Mandell JW. Fractalkine is a "find-me" signal released by neurons undergoing ethanol-induced apoptosis. Front Cell Neurosci. 2014;8:360. Epub 2014 Nov 7 PubMed.
- Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun. 2012 May;26(4):607-16. Epub 2012 Jan 30 PubMed.
- Elmer BM, Estes ML, Barrow SL, McAllister AK. MHCI requires MEF2 transcription factors to negatively regulate synapse density during development and in disease. J Neurosci. 2013 Aug 21;33(34):13791-804. PubMed.
- Erion JR, Wosiski-Kuhn M, Dey A, Hao S, Davis CL, Pollock NK, Stranahan AM. Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. J Neurosci. 2014 Feb 12;34(7):2618-31. PubMed.
- Koshimizu H, Takao K, Matozaki T, Ohnishi H, Miyakawa T. Comprehensive behavioral analysis of cluster of differentiation 47 knockout mice. PLoS One. 2014;9(2):e89584. Epub 2014 Feb 24 PubMed.
External Citations
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.