The Feud, Act II: Do Alzheimer’s Genes Affect Amyloid or Tau?
Quick Links
Most GWAS hits only nudge the risk of developing Alzheimer's; their real power lies in what they can tell researchers about how the disease develops. For some, scientists are starting to decipher how the proteins might influence pathology and—why, hello!—the old rivalry between Aβ and tau resurfaces. (In truth, the religious war of yesteryear lives on only as fodder for some media stories; most scientists agree both are important.) But consider this case in point: Previous work on BIN1 has linked the protein to tau pathology, not to amyloid. At the 12th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 18-22 in Nice, France, however,Taisuke Tomita of the University of Tokyo presented new data that tied both GWAS hits BIN1 and PICALM to Aβ production via endocytosis.
Tomita found that in mammalian cell cultures, each protein affected the endosomal recycling of one of the two secretases that produce Aβ, potentially boosting cleavage of the peptide. Meanwhile, poster presentations at AD/PD strengthened the case for BIN1 directly interacting with tau and exacerbating its toxicity, though the mechanism remains unclear. Some studies have correlated BIN1 expression with tau tangles in human brain, hinting that this interaction is relevant in the disease. However, more work is needed to elucidate how these proteins affect AD risk. If past tauist-baptist feuds are any guide, researchers may well find that the answer turns out to be neither one nor the other alone, but a bit of both.
BIN1 and PICALM were two of the first AD loci identified by GWAS, and they have popped up consistently in independent samples (see Apr 2011 news; Oct 2013 news). Risk variants in both genes correlate with a three- to six-month-earlier diagnosis of AD, suggesting they hasten pathology (see Nov 2014 news). Both proteins promote clathrin-mediated endocytosis, leading many researchers to speculate that they could alter the processing of endosomal amyloid precursor protein (APP) into Aβ, but hard data for this has been lacking (for review, see Bohm et al., 2015).

Interrupted Journey.
In control cells (left panel), most BACE (green) resides on the cell surface or travels to lysosomes for degradation, with only a little in early endosomes (purple). In cells lacking BIN1 (right panel), BACE builds up in endosomes (overlay appears white), where it can chop APP into Aβ. Bar, 10 μm. [Image courtesy of Taisuke Tomita.]
To pin down a mechanism, Tomita and colleague Takeshi Iwatsubo at the University of Tokyo turned to BIN1 knockout mice. BIN1 is an adaptor protein that helps pinch off endocytic vesicles. In the knockouts, Tomita saw higher levels of Aβ and BACE1. To find out what was happening to the secretase, he labeled cell-surface BACE1 in cultured neurons from the knockouts and tracked its movement. After 30 minutes, BACE1 appeared in early endosomes, just as it did in wild-type cells. After 90 minutes, however, BACE1 in the wild types had traveled to late endosomes and lysosomes for degradation, while in the knockouts the enzyme fell back, remaining stuck in endosomes. Since most Aβ is produced in endosomes after sequential cleavage of APP by BACE1 and γ-secretase, detaining BACE1 here would likely pump up Aβ production, Tomita noted. In addition, BACE1 might be recycled back to the plasma membrane, starting the process all over. The data add teeth to the idea that BIN1 can exert a direct effect on Aβ production by mediating BACE1 trafficking to the lysosome, Tomita concluded. He had previously presented some of this work at an Oct 2013 BACE conference (see Dec 2013 conference news).
Tomita is currently examining alternative splicing variants that confer a heightened risk of the disease. Preliminary results hint that one variant, which lacks the C-terminus, might act as a dominant negative, lowering the activity of normal BIN1. Other variants lie outside the coding region and might lower expression of the protein, he said.
This contrasts with other studies of BIN1, which point to tau. Previously, Jean-Charles Lambert of Lille University, France, associated an AD risk allele of BIN1 with higher BIN1 expression and more tau pathology in human brains (see Chapuis et al., 2013). The findings dovetailed with another human postmortem study, in which higher BIN1 expression in several brain regions correlated with more tau tangles, but not with amyloid plaques. In that study, splicing of BIN1 in AD brains was shifted away from the longest isoform toward shorter isoforms (see Holler et al., 2014). In accord with this, missplicing of BIN1 occurs in myotonic dystrophy, a disease also characterized by tau tangles in the brain (see Fugier et al., 2011; Caillet-Boudin et al., 2014).
How might BIN1 affect tau? Lambert had previously reported a direct interaction between the proteins, as seen in pull-down assays (see Aug 2012 conference news). In Nice, he and first author Yoann Sottejeau extended these findings. In new studies, they combined pull-downs of different fragments of BIN1 and tau with nuclear magnetic resonance spectroscopy to analyze binding. This revealed that the SH3 domain of BIN1 binds the proline-rich domain of tau, at residues 211-231. Multiple phosphorylation sites associated with AD are located within the proline-rich domain, and could potentially affect this interaction, Sottejeau noted in his poster.
To parse how this interaction affects pathology, researchers are using flies. Previously, Lambert had reported that knocking down the BIN1 homolog Amphiphysin in a Drosophila tauopathy model made the flies healthier and more agile, in line with the human data suggesting that too much BIN1 worsens tau tangles (see Aug 2011 conference news). In Nice, however, Thomas Jahn of the German Cancer Research Center, Heidelberg, presented opposite results. In a poster, first author Nina Dräger described finding higher tau levels and a worsening phenotype after knockdown of Amphiphysin in tau model flies. This implied the adaptor protein helped control tau pathology. Moreover, expressing human tau in wild-type flies and neuronal cells stimulated Amphiphysin expression. This might represent a compensatory mechanism to modulate tau accumulation, the authors suggested. It is unclear why different fly models produced such divergent results. In contrast to the mammalian data, neither Lambert nor Jahn saw any effect of BIN1 knockdown on flies modeling Aβ pathology. However, Jahn noted that this finding does not contradict the cell culture results, as both fly models overexpressed the Aβ peptide, not APP, and thus were not designed to assess effects on cleavage.
What to make of these conflicting findings? “I think the data linking BIN1 to tau pathology are solid,” noted Steven Estus at the University of Kentucky, Lexington. “However, BIN1 has a number of interaction domains and several isoforms, so it could be involved in many processes.” Estus suggested it would be worthwhile to see if BIN1 polymorphisms turned up in other tauopathies, such as frontotemporal dementia or progressive supranuclear palsy. Tomita told Alzforum he is examining BIN1 mouse models for any effects on tau. He found no change in levels of endogenous mouse tau in BIN1 conditional knockouts. Next, he will cross the conditional knockouts with transgenic mice that model tauopathies.
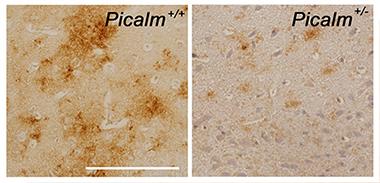
PICALM Promotes Amyloid Production.
Aβ (brown) accumulates in the piriform cortex of APP transgenic mice (left panel), but not in littermates that express half the normal level of PICALM (right panel). Bar, 100 μm. [Image courtesy of Taisuke Tomita.]
For PICALM, on the other hand, the data seem to point more clearly toward an amyloid link. An earlier study by researchers at Washington University in St. Louis reported that PICALM’s protein product, CALM, helps internalize APP, facilitating its cleavage into Aβ (see Xiao et al., 2012). Previous work by Tomita complemented that finding. He reported a lower ratio of Aβ42 to total Aβ in heterozygous PICALM knockout mice, which correlated with sluggish endocytosis of γ-secretase (see Kanatsu et al., 2014).
In Nice, Tomita presented new work in this area. To see if PICALM could affect amyloid pathology, he crossed PICALM heterozygous knockouts with transgenic mice that overexpressed APP. The offspring deposited about a quarter as much amyloid as the APP parent did, he told the audience. The researchers then introduced a mutation in the ANTH domain of CALM that weakened the protein’s ability to bind membrane lipids, which is crucial for promoting endocytosis. Overexpressing this mutant in cell culture lowered the Aβ42 ratio, just as lack of the protein did. This again tied the effect to altered endocytosis.
When CALM was mutated, more γ-secretase loitered on the cell surface. This also happened after knockdown of PICALM in wild-type cells. The data strengthen the idea that CALM helps internalize γ-secretase into endosomes, where it can produce Aβ, Tomita concluded. Next, he investigated how a rare variant of PICALM, I34M, affects this process. The SNP occurs in the lipid-binding site of the ANTH domain, and was reported as a possible protective variant. The researchers found that this variant poorly bound γ-secretase, stranding the enzyme on the cell surface. In these cells, the Aβ42 to total Aβ ratio dropped. Together, the PICALM and BIN1 data suggest that subtle effects on Aβ production can hasten or delay disease, Tomita told the audience in Nice.
“Thanks to its role in clathrin-mediated endocytosis, it makes sense to tie PICALM to APP processing, but the jury is still out on what exactly it’s doing,” Estus told Alzforum. He pointed out that PICALM plays a crucial role in keeping endocytosis running smoothly. Homozygous knockout mice die at weaning. For this reason, the protein probably would not be a tractable drug target, he noted.
Moreover, as with BIN1, PICALM has been linked to tau pathology. A 2014 paper from researchers at the University of Cambridge reported that CALM promoted tau clearance by stimulating autophagy. Both too much and too little CALM dialed down this waste-disposal system in fly and zebrafish models, leading to tau accumulation (see Moreau et al., 2014). Other studies are pursuing different mechanisms. PICALM is highly expressed in the vasculature, leading some researchers to speculate it might play a role in clearing Aβ from brain.—Madolyn Bowman Rogers
References
Alzpedia Citations
News Citations
- Large Genetic Analysis Pays Off With New AD Risk Genes
- Paper Alert: New Alzheimer’s Genes Published
- Do Alzheimer’s GWAS Hits Speed Up Disease?
- Meeting Explores Complex Biology of BACE Regulation
- GWAS Mega-Meta Yields More Risk Genes, BIN1 Binds Tau?
- Paris: President and All, French Science Takes the Stage
Paper Citations
- Bohm C, Chen F, Sevalle J, Qamar S, Dodd R, Li Y, Schmitt-Ulms G, Fraser PE, St George-Hyslop PH. Current and future implications of basic and translational research on amyloid-β peptide production and removal pathways. Mol Cell Neurosci. 2015 May;66(Pt A):3-11. Epub 2015 Mar 4 PubMed.
- Chapuis J, Hansmannel F, Gistelinck M, Mounier A, Van Cauwenberghe C, Kolen KV, Geller F, Sottejeau Y, Harold D, Dourlen P, Grenier-Boley B, Kamatani Y, Delepine B, Demiautte F, Zelenika D, Zommer N, Hamdane M, Bellenguez C, Dartigues JF, Hauw JJ, Letronne F, Ayral AM, Sleegers K, Schellens A, Broeck LV, Engelborghs S, De Deyn PP, Vandenberghe R, O'Donovan M, Owen M, Epelbaum J, Mercken M, Karran E, Bantscheff M, Drewes G, Joberty G, Campion D, Octave JN, Berr C, Lathrop M, Callaerts P, Mann D, Williams J, Buée L, Dewachter I, Van Broeckhoven C, Amouyel P, Moechars D, Dermaut B, Lambert JC, GERAD consortium. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry. 2013 Nov;18(11):1225-34. Epub 2013 Feb 12 PubMed.
- Holler CJ, Davis PR, Beckett TL, Platt TL, Webb RL, Head E, Murphy MP. Bridging integrator 1 (BIN1) protein expression increases in the Alzheimer's disease brain and correlates with neurofibrillary tangle pathology. J Alzheimers Dis. 2014;42(4):1221-7. PubMed.
- Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, Tosch V, Vignaud A, Ferry A, Messaddeq N, Kokunai Y, Tsuburaya R, de la Grange P, Dembele D, Francois V, Precigout G, Boulade-Ladame C, Hummel MC, Lopez de Munain A, Sergeant N, Laquerrière A, Thibault C, Deryckere F, Auboeuf D, Garcia L, Zimmermann P, Udd B, Schoser B, Takahashi MP, Nishino I, Bassez G, Laporte J, Furling D, Charlet-Berguerand N. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med. 2011 Jun;17(6):720-5. Epub 2011 May 29 PubMed.
- Caillet-Boudin ML, Fernandez-Gomez FJ, Tran H, Dhaenens CM, Buee L, Sergeant N. Brain pathology in myotonic dystrophy: when tauopathy meets spliceopathy and RNAopathy. Front Mol Neurosci. 2014 Jan 9;6:57. PubMed.
- Xiao Q, Gil SC, Yan P, Wang Y, Han S, Gonzales E, Perez R, Cirrito JR, Lee JM. Role of Phosphatidylinositol Clathrin Assembly Lymphoid-Myeloid Leukemia (PICALM) in Intracellular Amyloid Precursor Protein (APP) Processing and Amyloid Plaque Pathogenesis. J Biol Chem. 2012 Jun 15;287(25):21279-89. PubMed.
- Kanatsu K, Morohashi Y, Suzuki M, Kuroda H, Watanabe T, Tomita T, Iwatsubo T. Decreased CALM expression reduces Aβ42 to total Aβ ratio through clathrin-mediated endocytosis of γ-secretase. Nat Commun. 2014 Feb 28;5:3386. PubMed.
- Moreau K, Fleming A, Imarisio S, Lopez Ramirez A, Mercer JL, Jimenez-Sanchez M, Bento CF, Puri C, Zavodszky E, Siddiqi F, Lavau CP, Betton M, O'Kane CJ, Wechsler DS, Rubinsztein DC. PICALM modulates autophagy activity and tau accumulation. Nat Commun. 2014 Sep 22;5:4998. PubMed.
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.