Shooting Themselves in the Foot? Microglia Block “Good” State with ApoE4
Quick Links
Almost 30 years after its discovery as the most prominent risk factor for AD, ApoE continues to serve more questions than answers to the field. As evidenced by the many ApoE-focused presentations at this year’s Alzheimer’s Association International Conference, held July 31 to August 4 in San Diego, California, the brain’s deceptively humble lugger of lipids is still holding mechanistic surprises.
- ApoE4 made in microglia stymies these cells' protective responses to Aβ and tau pathology.
- In meninges, myeloid cells produce ApoE4, and lymphatic vessels expand in E4 mice.
- At the blood-brain barrier, ApoE4 triggers profound gene expression changes.
One theme focused on the old question of how ApoE4—the AD risk isoform—wreaks havoc in the brain. Researchers reported that when churned out by microglia, ApoE4 shuts down neuroprotective responses in the brain’s immune cells, thwarting their clearance of Aβ or tau pathology. Early findings hint that the lipids ApoE4 carries are poorly processed by lipoprotein lipase within microglia, potentially scrambling their metabolism and function. In the meninges, the primary producers of ApoE were found to be myeloid cells. In this triple-layered membrane sheath surrounding the brain, ApoE4 had the surprising effect of enlarging lymphatic vessels responsible for immune regulation and brain drainage. E4 also skewed the trajectory of age-related changes in cells that make up the brain’s vast blood-brain barrier, offering clues about how risk alleles might accelerate its erosion.
Although astrocytes produce the majority of ApoE in the brain, microglia can be provoked to churn out the apolipoprotein, too. In fact, ApoE is a critical component of microglial gene-expression signatures documented in the face of different neuropathological insults, including amyloidosis (Jun 2017 news; Sep 2017 news). Recent studies have found that the ApoE4 isoform exacerbates tau pathology and neurodegeneration in transgenic mice, but only when microglia are around (Oct 2019 news).
These and other findings cast microglia as the cells doing the damage at the behest of ApoE4, though they stop short of proving that microglia themselves are also the source of the ApoE4. In fact, a recent study led by David Holtzman at Washington University in St. Louis concluded that removal of ApoE4 only from astrocytes dampened microglial responses to tau, and partially quelled tau-induced neurodegeneration (Apr 2021 news).
At AAIC, Neta Rosenzweig, a postdoc in Oleg Butovsky’s lab at Brigham and Women’s Hospital in Boston, presented findings that directly addressed the question of what role microglial ApoE4 plays in AD. She used a menagerie of transgenic mice to investigate this question in different pathological contexts.
First, Rosenzweig riled microglia by injecting fluorescently labeled apoptotic neurons directly into the brains of ApoE3- or ApoE4-knock-in mice, which only express the human isoforms of the protein. Rosenzweig sorted out microglia that had gobbled up the fluorescent, dying neurons, looking at what genes they expressed. This showed her that while phagocytic microglia in ApoE3-KI mice had switched on the neurodegenerative expression profile, called MGnD, previously described by researchers in Butovsky’s group, those in ApoE4-KI mice failed to flip this transcriptional switch (Krasemann et al., 2017).
Marked by ramped-up expression of ApoE, TREM2, and a handful of other inflammatory and lipid metabolism genes, the MGnD profile resembles the disease-associated microglia (DAM) described by Ido Amit and Michal Schwartz at the Weizmann Institute of Science in Rehovot, Israel, where both Butovsky and, later, Rosenzweig, completed their graduate work (Keren-Shaul et al., 2017). One lingering question in the field has been whether these microglial transformations are beneficial or detrimental. In support of the former, Rosenzweig found that the MGnD-reticent ApoE4-KI microglia were also sluggish in approaching the injection site. Furthermore, they appeared inept at handling the debris they had consumed, as Rosenzweig spotted a backlog of internalized contents in their lysosomes.
Strikingly, none of these problems surfaced when Rosenzweig performed the same experiments in ApoE4-KI mice in which ApoE4 had been conditionally deleted from microglia. This suggested that ApoE4 produced by the microglia themselves was responsible for squelching productive responses.
Rosenzweig drew similar conclusions when she deployed this experimental paradigm in the APP/PS1 model of amyloidosis or in P301S-tau mice. In both transgenic mouse lines, each crossed to different ApoE-KI backgrounds, and microglia in ApoE4-KI mice failed to switch on the MGnD profile in response to the transgenic pathology—whether it was Aβ or tau. This exacerbated the accumulation of either of these pathologies in ApoE4-KI relative to ApoE3-KI mice. Conditional deletion of ApoE4 from microglia corrected these deficits, suggesting that ApoE4 produced by microglia, not astrocytes, was the source of the subpar microglial responses. In the APP/PS1 mice, Rosenzweig also tied microglial ApoE4 to poor recruitment of astrocytes to plaques. In the tau model, nixing ApoE4 from microglia even prevented neuronal loss.
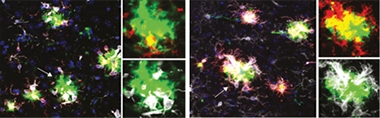
MGnD Repression Relieved. In APP/PS1 mice expressing human ApoE4 (left), microglia (white) around plaques (green) did not express the MGnD marker Clec7a (red). Removing ApoE4 from microglia only (right) restored microglial Clec7a and boosted recruitment to plaques. [Courtesy of Neta Rosenzweig, Brigham and Women’s Hospital, Boston.]
In all, the findings suggest that the MGnD transcriptional state is a beneficial one, and that ApoE4 production by microglia stymies the cells’ transition into it, rendering them less adept at handling neuropathology, Rosenzweig concluded.
To Butovsky, the finding that the MGnD state is a protective one in AD answers what has been a “Holy Grail” question in the field. However, that does not mean that this microglial response is always helpful. In fact, Butovsky and colleagues recently reported that in the context of glaucoma, a disease in which the ApoE4 genotype is protective, the microglial transition is a harmful one. The repression of MGnD by ApoE4 may explain why the isoform is protective in that disease (Margeta et al., 2022).
Jason Ulrich of Washington University called the findings consistent with previous work demonstrating a central role of microglia in ApoE4-related neurotoxicity. In the case of tauopathy, ApoE4 could influence the interplay between tau pathology, microglia, and neurodegeneration either by directly augmenting neurotoxic microglial responses, by somehow triggering the tau-related insult in neurons that leads to a neurotoxic microglial response, or a combination of these. “It will be important going forward to dig deeper into the different cell biological processes that microglia engage in their reactive state in the degenerating brain to determine which functions may be damaging and which may be protective,” Ulrich wrote to Alzforum.
Importantly, Rosenzweig spotted an echo of the dampened MGnD switch in ApoE4 carriers with AD. Using bulk RNA sequencing of microglia from postmortem brain samples, Rosenzweig found that compared to microglia in ApoE3/3 carriers, those in ApoE3/4 carriers expressed fewer genes from the MGnD playbook.
Of course, microglia in the human brain do not faithfully recapitulate MGnD, DAM, or any other mouse microglial profile (May 2019 news). The trend to name different glial gene-expression signatures—each detected in distinct experimental conditions, mouse models, or human disease states—recently prompted scientists across the field to call for a moratorium on christening such states with names, urging researchers to stick to detailed transcriptional and functional descriptions instead (Jul 2022 news).
One commonality between MGnD, DAM, and other previously reported disease-associated microglial states is the upregulation of genes involved in lipid handling, and these genes go far beyond ApoE. For example, they include lipid receptors such as TREM2, and the lipid hydrolyzer lipoprotein lipase (LPL). At AAIC, Kimberley Bruce of the University of Colorado in Aurora focused on LPL. This gene features prominently among the upregulated genes in microglia during development and in various disease states (Loving and Bruce, 2020). LPL is a secretory protein that, outside of the CNS, hydrolyzes triglycerides found in lipoproteins.
What it does in the CNS is less clear, as triglyceride-rich lipoproteins are typically rare there, Bruce said. However, she proposed that LPL could work in concert with lipoprotein receptors in the CNS to internalize all manner of lipid-laden debris. Myelin is one of them, as Bruce previously reported (Bruce et al., 2018). Genetic variants in LPL implicate this enzyme in AD, with loss-of-function variants upping risk, and gain-of-function variants reducing it (Bruce et al., 2020). Bruce previously reported that nixing LPL from microglia triggered the cells to transition into a pro-inflammatory state. Lipid droplets built up in them and cholesterol efflux plummeted, rendering the cells incompetent at processing Aβ, as well as lipids including myelin (Loving et al., 2021).
At AAIC, Bruce showed preliminary findings linking LPL to ApoE within microglia. First, she measured how efficiently LPL hydrolyzed lipids associated with different ApoE isoforms in vitro. When lipids were buddied up with ApoE4, LPL had a hard time processing them into fatty acids. A similar story played out in cultured microglial cells, where the cells poorly hydrolyzed lipids carried by ApoE4 relative to those carried by other ApoE isoforms. Bruce proposed that LPL is a critical modulator of microglial metabolism and inflammation. Together with ApoE, it plays a role in AD pathogenesis, she believes.
While Rosenzweig’s findings suggest a role for microglial ApoE in the brain parenchyma, other findings presented at AAIC hint that ApoE churned out by myeloid cells in the meninges may affect lymphatic drainage in that realm. Five years ago, scientists made the surprising discovery that the meninges—the triple layer of membranes that cradle the brain—house a lymphatic drainage system (Oct 2017 news). With age, the drainage becomes more stagnant (Jul 2018 news). The meninges also host a lively hub for immune cells, which enter and exit the brain via lymphatic vessels and, like satellites, communicate with immune cells residing in the brain parenchyma (May 2021 news).
At AAIC, Sandro Da Mesquita of the Mayo Clinic in Jacksonville, Florida, reported early findings about the role of ApoE this meningeal environment. Da Mesquita conducted some of his studies as a postdoc in Jonathan Kipnis’ group at Washington University in St. Louis.
Comparing the morphology of vessels crisscrossing the meninges in ApoE3-KI versus ApoE4-KI mice, Nikoleta Delivanoglou, a postdoc in Da Mesquita's lab, saw that, on average, lymphatic vessels in E4 mice were longer and wider than those in E3 mice, although Da Mesquita told Alzforum that some vessels in E4 mice retracted instead. ApoE knockouts had no expanded vessels, suggesting that the effect was due to a gain of function by ApoE4, Da Mesquita said. Notably, these E4-induced morphological changes were only significant in male mice. The researchers are currently investigating the mechanisms involved in this sexually dimorphic effect, and how the restructuring of meningeal vessels influences lymphatic drainage.
Using a spatial-transcriptomics technique called RNAScope to pinpoint which cells were expressing ApoE in the meninges, Da Mesquita pinned the vast majority of ApoE expression on resident macrophages there. Depleting these myeloid cells with a CSF-1R inhibitor drastically reduced expression of ApoE in the meninges.

In Meninges, Macrophages Express ApoE. Using RNAScope to image mRNA in the murine dura, macrophages (in green) can be seen expressing ApoE (red). Blood endothelial cells lining vessels express Pecam1 (blue). [Courtesy of Sandro Da Mesquita.]
Da Mesquita said that ongoing single-cell RNA-sequencing studies in the lab aim to decipher ApoE4-specific gene expression changes in these myeloid cells and other cell types in the meninges. Preliminary findings hint that macrophages in the meninges are more activated in ApoE4-KI mice, and that ApoE genotype more heavily sways the gene-expression profile of these macrophages than it does any other meningeal cell type.
Finally, according to studies led by Berislav Zlokovic, ApoE4 can promote the erosion of the blood-brain barrier. Previously, Zlokovic, University of Southern California, Los Angeles, had reported that with age, the blood-brain barrier becomes compromised, or leaky, in the hippocampi of ApoE4 carriers, and that this happens in a manner independent of Aβ or tau pathology (Jan 2019 news; May 2020 news). This degradation was fueled by activation of the CypA-MMP9 pathway in pericytes, which was detectable in the cerebrospinal fluid by way of the pericyte-injury marker sPDGFRb. (May 2012 news; Montagne et al., 2021).
At AAIC, Zlokovic presented fresh data on how ApoE4 alters gene expression in BBB-bolstering cells with age. Using single-nuclei RNA sequencing of brain endothelial cells and pericytes from the BBB, Zlokovic reported that different sets of genes in capillary endothelial cells and pericytes started to become dysregulated with age in ApoE4-KI mice relative to those in ApoE3-KI mice. The scientists saw differential expression of genes involved in cell junctions, cytoskeleton, and clathrin-mediated transport, as well as dramatic changes in the proteome and phospho-proteome—i.e., the landscape of phosphorylated residues among proteins. Together, these expression data implied that fundamental changes in BBB function were already afoot in relatively young, 2- to 3-month-old ApoE4-KI mice. These BBB changes preceded the onset of synaptic and memory deficits in 7-month-old ApoE4-KI mice relative to their ApoE3-KI counterparts, Zlokovic reported. He did not address which cells might have released the BBB-eroding ApoE4. However, he said that in ongoing experiments, researchers in his lab are testing how removal of ApoE from different cell types—including microglia and astrocytes—influences the effects on the barrier imparted by ApoE4.—Jessica Shugart
References
News Citations
- Hot DAM: Specific Microglia Engulf Plaques
- ApoE and Trem2 Flip a Microglial Switch in Neurodegenerative Disease
- In Tauopathy, ApoE Destroys Neurons Via Microglia
- Squelching ApoE in Astrocytes of Tau-Ravaged Mice Dampens Degeneration
- When It Comes to Alzheimer’s Disease, Do Human Microglia Even Give a DAM?
- Don’t Name It: Glial States Confound Easy Labels
- Lymphatic Vessels Found in Human Brain
- Lymphatic Brain Drain Withers in Aging, Worsens Disease
- As Mice Age, T Cells Traipse Around Their Meninges. Mayhem Ensues
- Absent Aβ, Blood-Brain Barrier Breakdown Predicts Cognitive Impairment
- Even Without Amyloid, ApoE4 Weakens Blood-Brain Barrier, Cognition
- ApoE4 Makes Blood Vessels Leak, Could Kick Off Brain Damage
Research Models Citations
Paper Citations
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O'Loughlin E, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Humulock Z, Zrzavy T, Conde-Sanroman P, Gacias M, Weng Z, Chen H, Tjon E, Mazaheri F, Hartmann K, Madi A, Ulrich JD, Glatzel M, Worthmann A, Heeren J, Budnik B, Lemere C, Ikezu T, Heppner FL, Litvak V, Holtzman DM, Lassmann H, Weiner HL, Ochando J, Haass C, Butovsky O. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity. 2017 Sep 19;47(3):566-581.e9. PubMed.
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I. A Unique Microglia Type Associated with Restricting Development of Alzheimer's Disease. Cell. 2017 Jun 15;169(7):1276-1290.e17. Epub 2017 Jun 8 PubMed.
- Margeta MA, Yin Z, Madore C, Pitts KM, Letcher SM, Tang J, Jiang S, Gauthier CD, Silveira SR, Schroeder CM, Lad EM, Proia AD, Tanzi RE, Holtzman DM, Krasemann S, Chen DF, Butovsky O. Apolipoprotein E4 impairs the response of neurodegenerative retinal microglia and prevents neuronal loss in glaucoma. Immunity. 2022 Sep 13;55(9):1627-1644.e7. Epub 2022 Aug 16 PubMed.
- Loving BA, Bruce KD. Lipid and Lipoprotein Metabolism in Microglia. Front Physiol. 2020;11:393. Epub 2020 Apr 28 PubMed.
- Bruce KD, Gorkhali S, Given K, Coates AM, Boyle KE, Macklin WB, Eckel RH. Lipoprotein Lipase Is a Feature of Alternatively-Activated Microglia and May Facilitate Lipid Uptake in the CNS During Demyelination. Front Mol Neurosci. 2018;11:57. Epub 2018 Mar 15 PubMed.
- D Bruce K, Tang M, Reigan P, H Eckel R. Genetic Variants of Lipoprotein Lipase and Regulatory Factors Associated with Alzheimer's Disease Risk. Int J Mol Sci. 2020 Nov 6;21(21) PubMed.
- Loving BA, Tang M, Neal MC, Gorkhali S, Murphy R, Eckel RH, Bruce KD. Lipoprotein Lipase Regulates Microglial Lipid Droplet Accumulation. Cells. 2021 Jan 20;10(2) PubMed.
- Montagne A, Nikolakopoulou AM, Huuskonen MT, Sagare AP, Lawson EJ, Lazic D, Rege SV, Grond A, Zuniga E, Barnes SR, Prince J, Sagare M, Hsu CJ, LaDu MJ, Jacobs RE, Zlokovic BV. APOE4 accelerates advanced-stage vascular and neurodegenerative disorder in old Alzheimer's mice via cyclophilin A independently of amyloid-β. Nat Aging. 2021 Jun;1(6):506-520. Epub 2021 Jun 14 PubMed.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
University of Arkansas for Medical Sciences
It is very intriguing to see so many studies documenting extensive differences in gene expression manifest as a function of APOE genotype. I would encourage anyone interested in this phenomenon to consider the growing body of data indicating that ApoE directly binds DNA in specific ways to act as a transcription factor (Theendakara et al., 2016). Most evidence indicates that it acts as a transcriptional repressor, well-documented through the competition between ApoE4 transcriptional activator TFEB at "CLEAR" sequences, cis elements that regulate genes necessary for apoptosis (Parcon et al., 2018; Lima et al., 2020). Recently, Islam et al. (2022) demonstrated that localization of ApoE to the nucleus is dramatically impacted by the presenilins.
Together, the data in this line of investigation provides a rational and compelling explanation for how ApoE4 will suppress autophagy—and perhaps related processes, such as LC3-associated endocytosis (Heckmann et al., 2019)—and thus contribute to the proteinopathy that now seems critical to the etiology of Alzheimer's disease.
References:
Theendakara V, Peters-Libeu CA, Spilman P, Poksay KS, Bredesen DE, Rao RV. Direct Transcriptional Effects of Apolipoprotein E. J Neurosci. 2016 Jan 20;36(3):685-700. PubMed.
Parcon PA, Balasubramaniam M, Ayyadevara S, Jones RA, Liu L, Shmookler Reis RJ, Barger SW, Mrak RE, Griffin WS. Apolipoprotein E4 inhibits autophagy gene products through direct, specific binding to CLEAR motifs. Alzheimers Dement. 2018 Feb;14(2):230-242. Epub 2017 Sep 22 PubMed.
Lima D, Hacke AC, Inaba J, Pessôa CA, Kerman K. Electrochemical detection of specific interactions between apolipoprotein E isoforms and DNA sequences related to Alzheimer's disease. Bioelectrochemistry. 2020 Jun;133:107447. Epub 2019 Dec 23 PubMed.
Islam S, Sun Y, Gao Y, Nakamura T, Noorani AA, Li T, Wong PC, Kimura N, Matsubara E, Kasuga K, Ikeuchi T, Tomita T, Zou K, Michikawa M. Presenilin Is Essential for ApoE Secretion, a Novel Role of Presenilin Involved in Alzheimer's Disease Pathogenesis. J Neurosci. 2022 Feb 23;42(8):1574-1586. Epub 2022 Jan 5 PubMed.
Heckmann BL, Teubner BJ, Tummers B, Boada-Romero E, Harris L, Yang M, Guy CS, Zakharenko SS, Green DR. LC3-Associated Endocytosis Facilitates β-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer's Disease. Cell. 2019 Jul 25;178(3):536-551.e14. Epub 2019 Jun 27 PubMed. Correction.
Make a Comment
To make a comment you must login or register.