On The Docket at AD/PD: The Many Crimes of ApoE4
Quick Links
Despite years of study, ApoE still has secrets to give up. At the 14th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 27–31 in Lisbon, Portugal, researchers indicted ApoE4 on several fronts. They charged it with holding back the microglial response to amyloidosis. They said it bungled disposal of damaged mitochondria, perhaps perturbing cellular energy metabolism. They implicated it in axonal remodeling, and said this may explain why the highly arborized neurons of the entorhinal cortex are particularly vulnerable to Alzheimer’s. Finally, as peripheral ApoE is beginning to attract more attention, scientists are accounting for the different pools of this lipoprotein that course through the human body. Others elaborated on how ApoE4 damages blood vessels in the brain and correlates with neuroinflammation. Quite the rap sheet.
- The APOE4 allele locks microglia in a homeostatic state.
- It impairs mitophagy and axonal plasticity.
- Peripheral APOE4 causes vascular problems in brain.
First, microglia, the “phenom” in AD research these days. Many genetic risk factors act through their effect on these cells (see Part 4 of this series), and some appear to be in cahoots with each other. Oleg Butovsky of Brigham and Women’s Hospital, Boston, previously reported that TREM2 and ApoE cooperate to rouse microglia into a “microglial neurodegenerative” (MGnD) state, in which they clean up dead neurons and other debris (Feb 2015 conference news; Sep 2017 news).
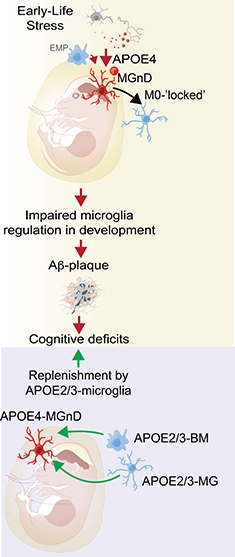
Microglial Replacement. ApoE4 microglia freeze into a homeostatic state; could replacing them with ApoE3 monocytes from blood restore normal function? [Courtesy of Oleg Butovsky.]
How does the ApoE4 allele affect this activation? In Lisbon, Butovsky said it has the same effect as knocking out ApoE, i.e., it traps microglia in a homeostatic state. His team isolated microglia from adult mice that carry humanized ApoE3 or ApoE4. In the former, microglia activated as the animal aged, but in the latter, homeostatic gene expression stayed high. Then the scientists challenged microglia by injecting dying neurons into mouse brain. Normally, this spurs microglia to enter the MGnD state. In E4 mice, however, it did not. They managed to turn down their homeostatic genes, but not to ramp up MGnD genes, like a teenager who can’t work up the motivation to get off the sofa and clean his room. Butovsky concluded that ApoE4 microglia are unable to respond properly to cellular damage in the brain.
In addition, aberrant behavior of ApoE4 microglia during development may render the brain vulnerable to Alzheimer’s, Butovsky believes. Charlotte Madore in his group found that microglia in ApoE3 mice assumed an MGnD phenotype during the first two weeks of life, when the cells are busy pruning inactive synapses. Then microglia settle into a calmer mode, turning up homeostatic genes. In newborn ApoE4 mice, however, microglia maintained high expression of homeostatic genes throughout infancy and into adulthood. Madore thinks this may prevent them from appropriately sculpting brain circuitry, and could lead to behavioral abnormalities later in life. Similarly, genetic ablation of TREM2 makes microglia “superhomeostatic” and prevents them from migrating toward tissue damage (May 2017 news). TREM2 knockout mice have abnormal social behavior as adults (Filipello et al., 2018; Calcagno et al., 2018).
Madore is investigating if these microglial differences interact with early life stress. Butovsky, working with Arie Kaffman at Yale University in New Haven, Connecticut, previously reported that early stress can cause lasting changes in microglia gene expression (Delpech et al., 2016). Madore found that stressors such as lack of bedding material cause mouse microglia to ramp up MGnD gene expression. If E4 mice cannot mount this response, perhaps that affects the brain in ways that leave mice susceptible to degeneration later, she speculated. In people, childhood epilepsy in ApoE4 carriers has been linked to an increased risk of AD late in life, hinting that early stressors could interact with genetic factors to predispose someone to the disease, Madore believes (Joutsa et al., 2017).
If ApoE4 microglia make the brain less able to combat AD, perhaps replacing them with healthier cells could be therapeutic, Butovsky suggested. In other studies, depleting brain microglia stimulated blood monocytes to enter the brain and repopulate the microglial niche (Lund et al., 2018; Cronk et al., 2018; Bennett et al., 2018). Microglia numbers can be reduced by preventing their proliferation with CSF1R blockers. Such drugs are under development for cancer, and treatment with one lessened pathology in a mouse model of AD (Sosna et al., 2018; Cannarile et al., 2017). Butovsky suggested infusing healthy ApoE3 monocytes into the bloodstream before CSF1R inhibitor treatment, to allow these cells to move into the brain and take over immune surveillance duties there. He is testing this idea in mouse models, collaborating with Li-Huei Tsai at MIT and Mathew Blurton-Jones at the University of California, Irvine.
ApoE affects other cellular processes that could play a role in AD, as well. Shira Simonovitch of Tel Aviv University, Israel, working with Daniel Michaelson and Ronit Pinkas-Kramarski there, previously reported that ApoE4 suppresses autophagy (Simonovitch et al., 2016). In Lisbon, Simonovitch extended this finding to mitochondrial disposal. The cell’s energy powerhouses are dynamic organelles that fuse and divide in response to stressors. Fusion boosts their health, while fission allows malfunctioning mitochondria to be chewed up by autophagosomes, a process known as mitophagy.
Simonovitch compared this in transgenic mice carrying human ApoE3 or ApoE4. In hippocampal sections from the E4 mice, Simonovitch counted three times as many elongated mitochondria as in E3 hippocampus. This hints at a block in fission, and staining for specific proteins revealed a dearth of the fission protein Drp-1 and an excess of the fusion protein Mfn1.
The scientists also saw that mitochondria in E4 brains had fewer crista, the folds of the inner membrane that produce energy, and western blots indicated changes in several known mitochondrial proteins. In particular, E4 mitochondria had a shortage of the cleaved form of pink1, which is associated with healthy mitochondrial resting potential. The data hint at an energy deficit in the ApoE4 brain; the increased mitochondrial fusion may be an attempt to compensate, Simonovitch suggested.
How about humans? The researchers have no way of measuring this directly in living people, but they compared postmortem samples from the brains of ApoE4/4 and ApoE3/3 AD patients. They found half as much Drp-1 in the former as the latter by western blot. Impairments in mitophagy have been linked to Parkinson’s and other neurodegenerative conditions (Aug 2013 news; Oct 2014 news; Sep 2016 news). Mitochondrial damage occurs in AD models, and removing these damaged organelles lessens pathology and improves memory in mouse and worm models of AD (Nov 2009 news; Feb 2010 news; Feb 2019 news).

Axonal Vulnerability? Entorhinal cortex neurons possess enormous axonal arbors (bottom right) that may leave them susceptible to tau pathology. [Courtesy of Tamamaki and Nojyo, 1993.]
ApoE4 also promotes tau pathology (Sep 2017 news). Neurons in the layer II entorhinal cortex (EC) and hippocampal CA1 region are particularly vulnerable to tau tangles, and Jean-Pierre Roussarie and colleagues at Rockefeller University wondered if ApoE isoforms might influence this. Roussarie works in the laboratory of Nobel laureate Paul Greengard, who passed away April 13 at age 93 (see obituary). Roussarie profiled seven different neuronal types in wild-type mice at three different ages to identify pathways most altered in vulnerable neurons compared with those that resist tau pathology. Together with Olga Troyanskaya’s group at Princeton University, Roussarie and colleagues then combined these expression profiles with a compendium of human genomics and GWAS data for genes associated with tau pathology (Greene et al., 2015; Beecham et al., 2014).
This pinpointed a tau vulnerability gene module that was highly connected in EC neurons, and whose gene members were suppressed during aging and in APPswe/PS1dE9 mice. Genes in this cluster regulate axon extension, microtubule assembly and transport, and neuronal signaling (Brichta et al., 2015; Roussarie et al., 2018). Importantly, gene-expression profiles from the different neuron types were comparable to those in postmortem human brain samples from the same regions, suggesting the results may translate to people, Roussarie said. This work is described in a paper posted to bioRχiv (Roussarie et al., 2018).
How about ApoE? Roussarie compared gene expression in healthy, year-old female ApoE2 and ApoE4 mice, and found 57 neuronal genes that were differently expressed in the E4s, about half turned up and the other half down. These genes overlapped extensively with the tau vulnerability module of axonal plasticity genes, as well as with genes associated with tau tangles in another GWAS (Chibnik et al., 2017). In other words, ApoE4 affects the same set of genes as do aging and AD pathology. Within EC neurons, the effects of ApoE4, aging, and tau pathology all converge on axonal plasticity, Roussarie concluded. He noted that EC neurons extend extremely complex axonal arbors, and speculated that this may leave them vulnerable to perturbations in this process.
While most studies of ApoE focus on the central nervous system, the protein is also abundant in the periphery. In Lisbon, Randy Bateman of Washington University in St. Louis reminded the audience that CNS and PNS versions are processed differently. In the CNS, all three isoforms turn over at the same rate, while in blood, they vary greatly. There, ApoE4 is cleared twice as fast as ApoE3. In line with this, people with one E3 and one E4 allele have more E3 than E4 in their plasma, while the opposite is true in brain (Wildsmith et al., 2012; Baker-Nigh et al., 2016).
To examine brain and plasma ApoE more closely, since then Bateman and colleagues have developed an assay to isolate ApoE from body fluids via monoclonal antibodies and analyze the protein by mass spectrometry. This taught them that, in cerebrospinal fluid, ApoE’s C-terminal domain was either missing or modified, Bateman reported in Lisbon. In addition, plasma and CSF ApoE differed at threonine 194. Though the researchers are still determining what this modification is, there are previously reported candidates at this location (Wernette-Hammond et al., 1989; Halim et al., 2013). These differences may help researchers design therapies to specifically target pathological ApoE, Bateman suggested.
Other research implicates peripheral ApoE in AD. Guojun Bu of the Mayo Clinic in Jacksonville, Florida, previously reported that peripheral ApoE4 makes brain blood vessels leaky and slows cerebral blood flow. This correlated with worse memory and more amyloid plaques in brain, even in animals without any CNS ApoE (Jul 2018 conference news). In Lisbon, Bu added to these data, reporting that blood vessels in mice with peripheral ApoE4 branch less than those in E3s and dilate less frequently, suggesting problems with regulating blood flow. E4s also deposited less collagen around blood vessels and had more astrogliosis in their brains.
To confirm that the peripheral ApoE4 effects also occur in wild-type animals that have CNS ApoE, Bu’s team injected plasma from young ApoE4 mice into aged wild-type mice, and found that it promoted leakage of the blood-brain barrier. ApoE3 plasma had the opposite effect, stemming leaks. “ApoE4’s harm likely comes from a combination of central and peripheral effects,” Bu concluded.—Madolyn Bowman Rogers
References
News Citations
- Parsing How Alzheimer’s Genetic Risk Works Through Microglia
- Microglia in Disease: Innocent Bystanders, or Agents of Destruction?
- ApoE and Trem2 Flip a Microglial Switch in Neurodegenerative Disease
- Paper Alert: TREM2 Crucial for Microglial Activation
- Parkinsonism-linked Protein Binds Parkin and Pink1, Drives Mitophagy
- ALS, Parkinson’s Proteins Co-Mingle in Mitochondria Destruction Pathway
- Gene Links Childhood Neurodegeneration to Mitophagy
- New Triple Transgenic Shows Mitochondrial Damage by Tau, Aβ
- Abnormal Mitochondrial Dynamics—Early Event in AD, PD?
- Could Disposing of Damaged Mitochondria Treat Alzheimer’s Disease?
- ApoE4 Makes All Things Tau Worse, From Beginning to End
- ApoE Has Hand in Alzheimer’s Beyond Aβ, Beyond the Brain
Paper Citations
- Filipello F, Morini R, Corradini I, Zerbi V, Canzi A, Michalski B, Erreni M, Markicevic M, Starvaggi-Cucuzza C, Otero K, Piccio L, Cignarella F, Perrucci F, Tamborini M, Genua M, Rajendran L, Menna E, Vetrano S, Fahnestock M, Paolicelli RC, Matteoli M. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity. 2018 May 15;48(5):979-991.e8. Epub 2018 May 8 PubMed.
- Calcagno N, Baufeld C, Madore C, Butovsky O. TREMendous 2 Be Social. Immunity. 2018 May 15;48(5):842-843. PubMed.
- Delpech JC, Wei L, Hao J, Yu X, Madore C, Butovsky O, Kaffman A. Early life stress perturbs the maturation of microglia in the developing hippocampus. Brain Behav Immun. 2016 Oct;57:79-93. Epub 2016 Jun 11 PubMed.
- Joutsa J, Rinne JO, Hermann B, Karrasch M, Anttinen A, Shinnar S, Sillanpää M. Association Between Childhood-Onset Epilepsy and Amyloid Burden 5 Decades Later. JAMA Neurol. 2017 May 1;74(5):583-590. PubMed.
- Lund H, Pieber M, Parsa R, Han J, Grommisch D, Ewing E, Kular L, Needhamsen M, Espinosa A, Nilsson E, Överby AK, Butovsky O, Jagodic M, Zhang XM, Harris RA. Competitive repopulation of an empty microglial niche yields functionally distinct subsets of microglia-like cells. Nat Commun. 2018 Nov 19;9(1):4845. PubMed.
- Cronk JC, Filiano AJ, Louveau A, Marin I, Marsh R, Ji E, Goldman DH, Smirnov I, Geraci N, Acton S, Overall CC, Kipnis J. Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J Exp Med. 2018 Jun 4;215(6):1627-1647. Epub 2018 Apr 11 PubMed.
- Bennett FC, Bennett ML, Yaqoob F, Mulinyawe SB, Grant GA, Hayden Gephart M, Plowey ED, Barres BA. A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron. 2018 Jun 27;98(6):1170-1183.e8. Epub 2018 May 31 PubMed.
- Sosna J, Philipp S, Albay R 3rd, Reyes-Ruiz JM, Baglietto-Vargas D, LaFerla FM, Glabe CG. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer's disease. Mol Neurodegener. 2018 Mar 1;13(1):11. PubMed.
- Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. 2017 Jul 18;5(1):53. PubMed.
- Simonovitch S, Schmukler E, Bespalko A, Iram T, Frenkel D, Holtzman DM, Masliah E, Michaelson DM, Pinkas-Kramarski R. Impaired Autophagy in APOE4 Astrocytes. J Alzheimers Dis. 2016;51(3):915-27. PubMed.
- Greene CS, Krishnan A, Wong AK, Ricciotti E, Zelaya RA, Himmelstein DS, Zhang R, Hartmann BM, Zaslavsky E, Sealfon SC, Chasman DI, FitzGerald GA, Dolinski K, Grosser T, Troyanskaya OG. Understanding multicellular function and disease with human tissue-specific networks. Nat Genet. 2015 Jun;47(6):569-76. Epub 2015 Apr 27 PubMed.
- Beecham GW, Hamilton K, Naj AC, Martin ER, Huentelman M, Myers AJ, Corneveaux JJ, Hardy J, Vonsattel JP, Younkin SG, Bennett DA, De Jager PL, Larson EB, Crane PK, Kamboh MI, Kofler JK, Mash DC, Duque L, Gilbert JR, Gwirtsman H, Buxbaum JD, Kramer P, Dickson DW, Farrer LA, Frosch MP, Ghetti B, Haines JL, Hyman BT, Kukull WA, Mayeux RP, Pericak-Vance MA, Schneider JA, Trojanowski JQ, Reiman EM, Alzheimer's Disease Genetics Consortium (ADGC), Schellenberg GD, Montine TJ. Genome-wide association meta-analysis of neuropathologic features of Alzheimer's disease and related dementias. PLoS Genet. 2014 Sep;10(9):e1004606. Epub 2014 Sep 4 PubMed.
- Brichta L, Shin W, Jackson-Lewis V, Blesa J, Yap EL, Walker Z, Zhang J, Roussarie JP, Alvarez MJ, Califano A, Przedborski S, Greengard P. Identification of neurodegenerative factors using translatome-regulatory network analysis. Nat Neurosci. 2015 Sep;18(9):1325-33. Epub 2015 Jul 27 PubMed.
- Roussarie JP, Yao V, Rodriguez-Rodriguez P, Oughtred R, Rust J, Plautz Z, Kasturia S, Albornoz C, Wang W, Schmidt EF, Dannenfelser R, Tadych A, Brichta L, Barnea-Cramer A, Heintz N, Hof PR, Heiman M, Dolinski K, Flajolet M, Troyanskaya OG, Greengard P. Selective Neuronal Vulnerability in Alzheimer's Disease: A Network-Based Analysis. Neuron. 2020 Sep 9;107(5):821-835.e12. Epub 2020 Jun 29 PubMed.
- Chibnik LB, White CC, Mukherjee S, Raj T, Yu L, Larson EB, Montine TJ, Keene CD, Sonnen J, Schneider JA, Crane PK, Shulman JM, Bennett DA, De Jager PL. Susceptibility to neurofibrillary tangles: role of the PTPRD locus and limited pleiotropy with other neuropathologies. Mol Psychiatry. 2017 Mar 21; PubMed.
- Wildsmith KR, Basak JM, Patterson BW, Pyatkivskyy Y, Kim J, Yarasheski KE, Wang JX, Mawuenyega KG, Jiang H, Parsadanian M, Yoon H, Kasten T, Sigurdson WC, Xiong C, Goate A, Holtzman DM, Bateman RJ. In vivo human apolipoprotein E isoform fractional turnover rates in the CNS. PLoS One. 2012;7(6):e38013. PubMed.
- Baker-Nigh AT, Mawuenyega KG, Bollinger JG, Ovod V, Kasten T, Franklin EE, Liao F, Jiang H, Holtzman D, Cairns NJ, Morris JC, Bateman RJ. Human Central Nervous System (CNS) ApoE Isoforms Are Increased by Age, Differentially Altered by Amyloidosis, and Relative Amounts Reversed in the CNS Compared with Plasma. J Biol Chem. 2016 Dec 30;291(53):27204-27218. Epub 2016 Oct 28 PubMed.
- Wernette-Hammond ME, Lauer SJ, Corsini A, Walker D, Taylor JM, Rall SC Jr. Glycosylation of human apolipoprotein E. The carbohydrate attachment site is threonine 194. J Biol Chem. 1989 May 25;264(15):9094-101. PubMed.
- Halim A, Rüetschi U, Larson G, Nilsson J. LC-MS/MS characterization of O-glycosylation sites and glycan structures of human cerebrospinal fluid glycoproteins. J Proteome Res. 2013 Feb 1;12(2):573-84. Epub 2013 Jan 11 PubMed.
Other Citations
External Citations
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
University of Arkansas for Medical Sciences
Regarding ApoE and autophagy, it appears the mechanism is a surprising one. A few years ago, Dale Bredesen and colleagues strengthened the evidence for ApoE being found intracellularly, in the nucleus and bound to DNA, in fact. As it turns out, the DNA sequence that Theendakara et al. found bound by ApoE4 is a CLEAR sequence, an enhancer bound by transcription factor EB (TFEB), a master regulator of autophagy genes.
References:
Theendakara V, Peters-Libeu CA, Spilman P, Poksay KS, Bredesen DE, Rao RV. Direct Transcriptional Effects of Apolipoprotein E. J Neurosci. 2016 Jan 20;36(3):685-700. PubMed.
Parcon PA, Balasubramaniam M, Ayyadevara S, Jones RA, Liu L, Shmookler Reis RJ, Barger SW, Mrak RE, Griffin WS. Apolipoprotein E4 inhibits autophagy gene products through direct, specific binding to CLEAR motifs. Alzheimers Dement. 2018 Feb;14(2):230-242. Epub 2017 Sep 22 PubMed.
Universities of Manchester and Oxford
APOE-ε4 is certainly not always the villain, although it is so in relation to HSV1 and AD and to herpes labialis, and to HSV2 in the case of genital herpes. It can be the hero in other diseases. A case in point is its protective action against damage induced in liver by hepatitis C virus (Wozniak et al., 2002; Gong and Cun, 2019).
We investigated this disease, and various others known to be caused by infectious agents, to find if APOE plays a possible role in their occurrence or severity, selecting a disease on the criterion that the microbe responsible bound to the same receptors in the cell surface, such as heparan sulphate proteoglycans (HSPG), as did ApoE isoforms. The rationale was that if one specific isoform bound less strongly than the others, it would allow greater cell entry of the microbe and hence more damage. The data we obtained revealed a modulatory effect of APOE in various microbial diseases, with the HCV results being the strongest because of the larger number of well-characterized samples available (Itzhaki and Wozniak, 2009). We argued that from an evolutionary point of view, APOE-ε4 might have conferred an advantage in protecting against certain pathogens such as HCV, rather than being selected against because of its conferring susceptibility to AD and atherosclerosis.
References:
Wozniak MA, Itzhaki RF, Faragher EB, James MW, Ryder SD, Irving WL, . Apolipoprotein E-epsilon 4 protects against severe liver disease caused by hepatitis C virus. Hepatology. 2002 Aug;36(2):456-63. PubMed.
Gong Y, Cun W. The Role of ApoE in HCV Infection and Comorbidity. Int J Mol Sci. 2019 Apr 25;20(8) PubMed.
Itzhaki RF, Wozniak MA. Apolipoprotein E: Microbial Friend or Foe?. In: Apoprotein Research, Eds: LR Penfield and RT Nelson 2009. Nova Science Publishers Inc., New York.
Make a Comment
To make a comment you must login or register.