Past Webinar
How Does Excess Aβ Leave the Brain, How Does It Get In, And Can We Trap It Outside?
Quick Links
Introduction
Berislav Zlokovic led this live discussion on 23 June 2003. Readers are invited to submit additional comments by using our Comments form at the bottom of the page.
Transcript:
Berislav Zlokovic, University of Rochester, led this live discussion on 23 June 2003.
Participants: Kim Green; Ladislav Volicer, Boston University Medical School; Berislav Zlokovic, University of Rochester Medical Center, New York; Martha E, Stokely University of North Texas Health Sciences Center; Dave Holtzman, Washington University, St. Louis, Missouri; Alexei Koudinov, Peoples Friendship University of Russia, Moscow; Anne Fagan, Washington University in St. Louis; Yasuji Matsuoka, New York University School of Medicine; Jorge Busciglio, University of Connecticut Health Center; Rolf W, Warzok University of Greifswald, Bonn, Germany; Tadafumi Hashimoto, University of Tokyo; Andrea, Case Western Reserve University; Leigh Holcomb, Texas A&M Neuropsychiatry Research Program; Gaku Sakaguchi, Nathan Kline Institute; Dietmar Thal, University of Bonn Medical Center, Germany; Atul Deshpande, University of Connecticut Health Center; Michal Schnaider, Beeri Psychiatry Department at Mount Sinai School of Medicine.
Note: The transcript has been edited for clarity and accuracy.
Gabrielle
Hi, and welcome everyone. I am Gabrielle Strobel, managing editor of Alzforum, and pleased to moderate today. While people are still arriving, perhaps Berislav could start with a question growing right out of his Nature Medicine paper. Many people probably wonder if soluble Receptor for Advanced Glycation End-products (sRAGE) could be a potential therapy, Berislav?
Berislav Zlokovic
In principle, Ab-binding agents could be, and sRAGE is part of that group. So the answer is yes.
Dave Holtzman
Berislav, is it known where sRAGE is metabolized systemically after it is injected?
Berislav Zlokovic
The pharmacokinetic studies are in progress; it is likely to be excreted by the kidney and perhaps liver.
Yasuji Matsuoka
Are there any compounds with an affinity to the physiological form of Ab that could be sequesterers?
Berislav Zlokovic
The higher the affinity, the better.
Yasuji Matsuoka
Berislav, any Ab deposition in peripheral organs after treatment?
Berislav Zlokovic
Not observed, but we did not look hard enough into it.
Gabrielle
Perhaps we could address the first question in the backgrounder: What should the next experiments be to advance this potential therapeutic approach?
Berislav Zlokovic
Perhaps to look into high affinity Ab peripheral binding agents that could also reduce inflammation and improve cerebral blood flow (CBF). Possibly further chemical design of the sRAGE molecule to optimize its binding to Ab, and producing smaller molecules, such as V-domains.
Kim Green
As RAGE is transporting Ab into the brain, would this suggest that Ab is being produced in the periphery?
Berislav Zlokovic
Ab can be produced in the periphery, but also during processes that dissolve amyloid plaques from the brain, so a bulk may come from the CNS.
Kim Green
So you would surmise that increased cerebral AbP leads to decreased CBF?
Berislav Zlokovic
Yes, increased Ab decreased CBF. After treatment with sRAGE, the levels go up from normal 50-100 pmol/L to about 1.5 nmol/L, about a 20-fold increase.
Kim Green
Interesting, as I would guess that a decrease in CBF would cause an increase of AbP!
Berislav Zlokovic
Kim, what exactly is AbP?
Kim Green
Amyloid b peptide production.
Berislav Zlokovic
We did not look into it; I was referring to Ab levels.
Alexei Koudinov
Berislav, add (with regard to the possibility of the systemic production of Ab) that we showed some years ago that Ab is produced in HepG2 hepatocytes (Cell Biol Inter, 1997).
Dave Holtzman
How far along are experiments to determine whether RAGE KO x APP transgenic mice exhibit changes in Ab deposition/Ab metabolism?
Berislav Zlokovic
Some of it has been completed, but only in the earlier age group, five to six months, I believe. They do much better on behavioral tasks and develop less Ab. More conclusive studies are in progress with older age groups.
Yasuji Matsuoka
Berislav, what APP mice did you cross with?
Berislav Zlokovic
With RAGE KO. This work has been done with Shid Du Yan and Mark Kindy. The APP was PD-hAPP.
Gabrielle
Berislav, did you do your sRAGE treatments in different mouse models that varied in terms of how much cerebral amyloid angiopathy (CAA) they had? I wonder what your data reveal about CAA.
Berislav Zlokovic
Detailed studies are in progress. In terms of CAA, we showed that sRAGE treatment increases blood flow.
Yasuji Matsuoka
Berislav, any notable amount of peripheral Ab in PDAPP?
Jorge Busciglio
Berislav, I have a basic question: Is RAGE a receptor for the monomeric, oligomeric or fibrillar form of Ab?
Berislav Zlokovic
According to literature, it can bind both fibrillar and monomeric forms.
Dave Holtzman
Berislav, while it seems likely from your paper that the effect on Ab deposition of chronic sRAGE treatment of APP mice over three months is due to a peripheral effect, can you rule out a central effect of the small percentage of sRAGE that crosses into the CNS?
Gabrielle
While Berislav is answering some questions put to him, I was wondering if Yasuji could tell us about ongoing work with small molecules that bind Ab in the periphery. Anything you could tell following your paper on gelsolin?
Yasuji Matsuoka
We are testing other classes of Ab binding agents because chemical modification of gelsolin and GM1 is not realistic. I found a few Ab binding agents that could alter both brain and plasma Ab.
Gabrielle
All: Do receptor-mediated transport mechanisms respond in the same way to peripheral sequestration as passive flow would, i.e., will CNS levels go down if Ab is removed from the peripheral pool?
Berislav Zlokovic
I think that capturing Ab in the periphery will lower the total amount of Ab from the peripheral pool available for exchange with its central pool; this might move Ab from brain to periphery.
Alexei Koudinov
Dr. Zlokovic should be an expert in this subject with his pioneering earlier papers on receptor-mediated Ab transport. I mean receptors related to lipoprotein (LP) transport.
Berislav Zlokovic
Alexei, we believe that LRP-1 is important in elimination from brain.
Tadafumi Hashimoto
Berislav, is LRP-1 the only one?
Alexei Koudinov
...and this Q points to a pre-discussion background point on AD as a systemic disease. If so, the change in the systemic pool of Ab should affect the CNS levels.
Berislav Zlokovic
We found that LRP-1 clusters II and IV bind Ab and its mutants with high affinity. It is probably a major efflux mechanism at the BBB. There may be some other mechanisms, depending on which form Ab is in.
Dietmar Thal
Dr. Zlokovic, what do you think is the role of astrocytes in regulating Ab homoeostasis in the neuropil, especially in regard to their function in the blood brain barrier?
Berislav Zlokovic
Dietmar, I am not sure that the regulatory role of astrocytes in Ab BBB transport is well-understood at this time. Very interesting possibility. Perhaps Dave can make some comments regarding GFAP-ApoE mice.
Dave Holtzman
I think it is likely that ApoE regulates BBB transport of Ab. ApoE is made in astrocytes. Whether astrocytes play a role outside of ApoE and also ApoJ production, I don't think anyone knows.
Gabrielle
Rolf Warzok has worked on the p-glycoprotein (known as the transporter that pumps drugs out of tumor cells), suggesting it may transport Ab out of endothelial cells, as well. Do we know anything about its role vs. LRP? Berislav, all?
Rolf W. Warzok
In our studies we found an inverse correlation between P-glycoprotein and Ab load. Subjects with ApoEE4 had lowest PGP levels.
Gaku Sakaguchi
Does LRP-1 have a sensor machinery for decreasing amyloid in the periphery?
Berislav Zlokovic
Hi, Gaku; yes, it has high affinity to bind Ab in periphery as well, and soluble fragments may act as binding agents.
Dave Holtzman
In regard to Ab in the blood, if it is bound to a large molecule, that should prevent it from reentering the brain unless the large molecule is actively transported into brain. If there are transport systems that "sense" Ab levels in the periphery and somehow respond to that in some way, it is not known, I believe.
Berislav Zlokovic
I agree with Dave.
Gabrielle
Again, while Berislav is busy replying, can I ask Dave and Yasuji one of the questions in the background text? Is serum Ab truly out of the picture once it is bound? Could transport systems "sense" that bound serum Ab is increasing, and reduce Ab efflux in response? If so, peripheral sequestration could lead to an unintended increase of CNS Ab. Have animal models ruled this out? Or is there an error in the thought?
Yasuji Matsuoka
Plasma Ab levels returned to the baseline quickly after treatment with simple Ab binding agents, such as gelsolin, GM1 and new testing compounds. Ab disappeared from the blood in the case of simple Ab binding agents. We are working out the pathway.
Atul Deshpande
Recently there was an article suggesting that astrocytes internalize Ab. Would that be of any significance in decreasing the Ab load in the brain and transporting it across the BBB into the blood? (See ARF related news story).
Dave Holtzman
It would seem that astrocytes have the potential to play a major role in Ab metabolism, since they express many receptors for molecules that bind Ab.
Alexei Koudinov
Dave, that's what I meant by saying earlier that Ab transport regulation is a systemic event, the sensing machinery of transport.
Dave Holtzman
Alexei, I agree with you that it could be a "systemic" event.
Berislav Zlokovic
If astrocytes can resolubilize amyloid and release free Ab, in the presence of a stable efflux system at the BBB, this can be a possibility. I am not aware, however, of any study that has tested this possibility in an animal model.
Gabrielle
Another question for all from the background text: Are there ways of stimulating peripheral degradation of Ab in the liver and the kidney? That would take it out of the equation faster, too.
Berislav Zlokovic
Yes, I totally agree—same as dialysis.
Dietmar Thal
What is the role of ApoE in regard to the drainage of Ab from the brain to the vessels? Do you have an explanation for our finding that capillary Ab deposition is strongly associated to the ApoE4 allele?
Alexei Koudinov
Dietmar, what is your explanation for this event?
Berislav Zlokovic
We are studying details now. In our earlier work (J Neurochem, 1997), we showed that ApoE2 and 3 prevent Ab transport across the BBB, while ApoE4-bound Ab can get into the brain from periphery.
Dietmar Thal
Alexei, our attempt to explain this finding is that Ab-ApoE complexes are less soluble when E4 is present.
Dave Holtzman
It is very interesting that one of the major effects of ApoE on Ab is on CAA (even more than on parenchymal plaques). Perhaps drainage of Ab from brain and/or via the BBB is influenced by the transport/drainage of ApoE out of brain via receptors/heparin sulfate proteoglycans (HSPGs) and others.
Berislav Zlokovic
Dave, I agree; this is an interesting possibility. It could also be related, though, to the level of LRP-1 expression at the BBB.
Leigh Holcomb
Could you explain how HSPG would be involved?
Dave Holtzman
HSPGs are concentrated near vessels, and ApoE binds strongly to HSPG. I agree LRP-1 may also help co-localize ApoE at the BBB.
Berislav Zlokovic
This is also a possibility, as ApoE binds to clusters II and IV as well as Ab.
Gabrielle
Berislav, for the non-expert: What are clusters II and IV?
Berislav Zlokovic
These are soluble forms of LRP-1 receptors part II and IV. (There is VI + cytoplasmic tail.)
Alexei Koudinov
Dietmar, I remember that we discussed while referring to and discussing papers by Berislav that there could be a competition for Ab and ApoE (E2-, 3-, 4-specific) for binding to LPR.
Dietmar Thal
Are there any other pathological alterations in blood vessels, besides CAA, that could be responsible for lowering Ab resorption?
Berislav Zlokovic
Yes, this includes senescence of the vascular system (replicative or stress-induced) and aberrant angiogenesis in response to VEGF and bFGF.
Gabrielle
Berislav, Yasuji, Dave, others, are you trying to find and develop lead compounds on your own, or have companies licensed the drug development piece? I am asking because Alzforum is thinking about what sorts of information resources we could help establish for academic scientists who are pursuing their treatment hypothesis themselves, at least part of the way, to create more validated drug leads. What sorts of information can be hard to find that we could help assemble? Contacts and names of contract research organizations (CROs) who do toxicology and pharmacokinetic studies? Places to purchase compound libraries? Any needs we should think about?
Yasuji Matsuoka
Gabrielle, yes, I am trying.
Rolf W. Warzok
Dietmar, vessels with high Ab have low PGP and vice versa. We never registered a coexpression of Ab and PGP in the same vessels. In other words, only in vessels with no PGP was Ab observed in double staining immunohistochemistry.
Gabrielle
Interesting; so you suspect PGP to be a major exporter? How about LRP? Did you assess that?
Dave Holtzman
If PGP is a major exporter, wouldn't you expect PGP KO mice to have higher levels of Ab in the CNS? Has this been done?
Rolf W. Warzok
So far we haven't looked at LRP. We are just now looking at this in mice.
Berislav Zlokovic
Rolf, we were not able to observe so reproducibly that ABC transporters are associated with CAA using microarray analysis, but in about 50-60 percent of AD cases we also found downregulation of ABA-1 associated with Tangier's disease, and MRP-1 associated protein. We also reported that reduced expression of LRP-1 is associated with increased levels of vascular Ab. The CAA here may also be secondary to senescence, and Ab accumulation secondary to LRP-1 downregulation that is down in senescent endothelium.
Atul Deshpande
In AD, due to inflammation and activation, the BBB is probably compromised; that could also play a role in the entry of Ab into the brain and vice versa.... Just a thought.
Gabrielle
Also, Berislav, do you know the effect of RAGE deficiency on the steady-state endogenous Ab levels in the brain and plasma? Someone asked that in a comment.
Berislav Zlokovic
It's in works currently with Shi Du Yan from Columbia.
Gabrielle
We are nearing the end of the hour. Let me thank Berislav and everyone very much for coming. This looks like a promising line of investigation and we sure hope something comes of it.
Background
Background Text
By Berislav Zlokovic
In yesterday's online version of Nature Medicine, our lab and colleagues published a report about blocking Ab brain import from the periphery across the blood-brain barrier via the endothelial receptor RAGE. The text below first summarizes the main news in this paper, and then lays out the transport-clearance hypothesis of Ab and related Ab-lowering strategies in more breadth and detail.
Synposis of Deane et al. (15a)
We found that RAGE in brain endothelium mediates transport of circulating unbound (free) Ab across the blood-brain barrier (BBB). At pathophysiological levels, this transport results in neurovascular stress and reductions in cerebral blood flow (CBF). Both a soluble form of RAGE (sRAGE) and RAGE-specific IgG block Ab transport at the BBB and the resulting reductions in CBF in wild-type and Tg2576 mice. Treatment of PD-hAbPP mice with sRAGE reduces amyloid load and Ab levels in the brain. Data suggest that peripheral non-immune scavenging agents such as sRAGE efficiently shift Ab exchanges across the BBB, favoring egress of peptide from brain. Flux calculations indicate that at pathophysiological Ab plasma levels in Tg2576 mice or PD-hAbPP mice treated with an antibody or sRAGE, the Ab plasma pool can still rapidly replenish Ab brain levels at remarkably high rates close to 0.15 micromoles/kg brain interstitial fluid per day. Thus, neutralizing the peripheral pool of Ab and blocking its transport across the BBB should lower the risk of generalized neuroinflammation and of compromise of the blood flow during Ab-lowering therapeutic interventions associated with increases in circulating free Ab.
Ab Transport-Clearance Hypothesis
Increases in Ab production can explain a small percentage of early-onset cases of familial AD in those people who carry inherited mutations in the AbPP gene that flank the Ab coding region (i.e., Swedish mutation) or the presenilin 1 or 2 genes. (27) Increases in production have not, however, been found in sporadic AD, or in familial AD/CAA where people inherited mutations inside the Ab coding region (e.g., Dutch, Iowa); an exception here is the Flemish mutation. Consequently, we could regard b-amyloidosis in sporadic and even some familial AD as a "storage" disease caused by inefficient clearance of a peptide that is normally produced in the CNS. (28,36,37) In general, any mismatch between Ab production, transport of circulating Ab into the CNS and clearance-whether resulting from increased production, increased transport of blood-borne peptide or inadequate clearance-may result in Ab accumulation in the CNS, and the two plausible hypotheses for its clearance from the brain are metabolism (41,42,89) and transport out. (36,37) Here we will discuss Ab transport, and why our current understanding of this process lends support for the therapeutic strategy of the "peripheral sink."
Transport of Ab in the CNS: Part Drifting, Part Shipping
Nonspecific bulk flow of brain interstitial fluid seems to be responsible for about 10-15 percent of Ab clearance from normal brain. (29) The blood-brain-barrier (BBB) normally prohibits free exchange of polar solutes, such as Ab, between brain and blood or blood and brain, mostly because of the presence of tight junctions between brain endothelial cells that form a continuous monolayer. Therefore, carrier-mediated or receptor-mediated transport system(s) for Ab must exist at the BBB to remove Ab from the CNS and inject it into circulation shortly after its physiological production (37), or to shuttle circulating Ab into the CNS. In 1993 we suggested that carrier and/or receptor-mediated transport across the BBB regulates brain Ab (34), and since then numerous reports from different groups have verified this hypothesis. (1-5,8,9,11,16-23,25,26,29,34-37)
1. Brain Export
Recent studies in PDAbPP mice have demonstrated that a single intravenous injection of the m266 monoclonal anti-Ab antibody promotes a rapid outflow of Ab from the CNS into plasma, increasing plasma Ab from baseline levels of 200 pg/ml to 5-10 ng/ml within 24 hours. (3) Given the similarity in plasma and CSF levels of Ab between humans and PDAbPP mice (6), DeMattos et al. suggested that Ab efflux measurements from brain to plasma after challenge with an anti-Ab antibody may be useful for quantifying brain amyloid burden in patients at risk for Alzheimer's, or diagnosed with the disease (see ARF related news story). As plaques develop in primate models of b-amyloidosis and in transgenic mice, soluble Ab from brain and plasma settles onto amyloid deposits in the CNS and around blood vessels and, consequently, the transport equilibrium for Ab between the CNS and plasma shifts (1,3-5,18). Using a squirrel monkey model of cerebral amyloid angiopathy (CAA), our lab recently confirmed that Ab is rapidly eliminated from brain into plasma across the BBB, and we noticed an age-dependent decline in this Ab clearance via the BBB that correlates with increases in amyloid deposition and Ab cerebrovascular immunoreactivity. (1,18)
How Does Ab Get Out? Ask LRP-1
LRP-1 is a large, multifunctional scavenger and signaling receptor belonging to the LDL receptor family. (10) LRP-1 was first discovered as a key endocytic receptor for the transport and metabolism of cholesterol and ApoE-containing lipoproteins. Its 515 kDa heavy chain contains four ligand-binding domains (clusters I-IV) that bind numerous structurally unrelated ligands, including ApoE, a-macroglobulin, tissue plasminogen activator, plasminogen activator inhibitor-1, AbPP, factor VIII, and lactoferrin. The 85 kDa light chain of LRP-1 contains a transmembrane domain and a cytoplasmic tail. The latter can be phosphorylated on serine, which has been linked to enhanced endocytosis, or on tyrosine; see review. (10)
We recently showed that LRP-1 functions as a clearance receptor for Ab at the BBB. (29) The LRP-1-mediated crossing (or transcytosis) of Ab begins at the brain side of the endothelium and is, therefore, directly responsible for eliminating Ab from the brain's interstitial fluid into blood (Fig. 1). It is not understood what the exact molecular mechanisms are that regulate interactions of Ab?with LRP-1 at the BBB, but we do know that the LRP-1 ligands ApoE and a-macroglobulin can influence Ab clearance.
The expression of brain endothelial LRP-1 appears to go down during normal aging in rodents, nonhuman primates, and in AD patients associated with positive staining of vessels for Ab40 and Ab42. (29) There is a genetic association between LRP-1 and the development of AD, but the biochemical mechanisms by which LRP-1 could affect the onset of the disease remain unknown. (10) Our most recent in vitro surface plasmon resonance studies indicate that Ab40 and Ab42, as well as Dutch and Dutch/Iowa mutants of Ab40, are direct ligands for sLRP-1 clusters II and IV; moreover, all Ab peptides bind directly to the abluminal site of the BBB via LRP-1 with high affinity. (38)
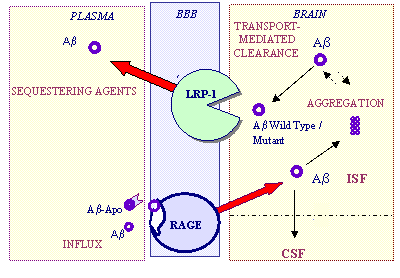
2. Brain Import
The autosomal dominant mutations that cause early-onset AD all increase Ab42 in plasma and brain. A late-onset AD locus on chromosome 10 acts to increase plasma Ab. The few studies that have analyzed plasma Ab levels in AD patients vs. age-matched controls suggest either no change or increased levels in AD, and/or an increased risk for AD in cognitively normal elderly individuals with high levels of plasma Ab as reviewed. (6) Tg2576 mice overexpressing AbPP develop high plasma levels of Ab40 and 42 (4 nM and 0.5 nM, respectively), between three and nine months of age. (14) PDAbPP mice have significantly lower baseline values of plasma Ab (~200 pg/ml), but these levels shoot up 40-fold after a single intravenous injection of anti-Ab antibody (3) or sRAGE. (33). Ab circulating at such pathophysiological concentrations can be rapidly transported back into the CNS. (11,15,20,34) Therefore, trapping Ab in plasma is critical to reducing Ab levels in the CNS and to shift the plasma-CNS equilibrium of Ab, at least in these models.
Agents that bind Ab in plasma but do not themselves penetrate the BBB may promote the outflow of a rapidly mobilized, soluble pool of Ab, acting as a peripheral "sink." If such agents do enter the CNS, they may bind soluble brain Ab. This would promote resolubilization of previously aggregated Ab as the brain equilibrium between soluble and aggregated Ab shifts towards the soluble side, and this in turn should result in Ab elimination from the CNS provided the clearance systems are intact.
How Does Ab Get In? Ask RAGE
RAGE is a receptor in the immunoglobulin superfamily. In addition to Ab, it binds a broad repertoire of ligands, including products of nonenzymatic glycoxidation (AGE), the S100/calgranulin family of proinflammatory cytokine-like mediators, and the high-mobility group 1 DNA-binding protein amphoterin. (31) RAGE biology is largely dictated by its ligands in that mature animals show little RAGE expression in most tissues until deposition of ligands triggers expression. When pathogenic Ab species accumulate in AD (32) or transgenic models of b-amyloidosis, RAGE expression increases in affected cerebral vessels, neurons, or microglia. In contrast to the ligand-mediated receptor downregulation observed with LDL receptors in a lipoprotein-rich environment (10), or LRP-1 in an Ab-rich environment (29), RAGE is upregulated by its ligands. This mechanism could exacerbate cellular dysfunction. RAGE binds soluble Ab in the nanomolar range, and then mediates pathophysiologic cellular responses. (31) RAGE is upregulated in the AD brain vasculature (32) and it regulates binding and transport of Ab in a human model of the BBB. (16) In light of these findings, we have recently shown that, in vivo, Ab binding to RAGE at the brain endothelium may increase transport of circulating Ab into the CNS. We also think RAGE is involved in diminishing cerebral blood flow, accompanying amyloid angiopathy, and proinflammatory events as observed in AD brain. In support of this hypothesis, we have found that RAGE mediates transport of physiological and pathophysiological concentrations of plasma Ab across the BBB into the brain, and that the latter leads to expression of proinflammatory cytokines in neurovascular cells and elaboration of endothelin-1, causing decreased cerebral blood flow. (15,15a)
Finally, there is also LRP-2 at the BBB. It may import plasma Ab complexed with apolipoprotein J (ApoJ). (35) Yet LRP-2 is normally saturated by high levels of plasma ApoJ , which precludes significant influx of Ab into the CNS. This leaves RAGE as the major Ab influx receptor at the BBB.
Therapeutic Strategies
Where does all this leave us? Peripheral Ab-binding agents may well promote clearance of brain-derived Ab, thereby reducing Ab levels and amyloid load in the CNS of different AbPP-overexpressing mice. In the current online Nature Medicine, we have shown that sRAGE completely prevents transport of circulating Ab into the CNS, and since it does not penetrate the CNS in appreciable amounts it may act as a sink, favoring egress of Ab from brain. (15,15a) Moreover treatment with sRAGE improves the blood flow in Tg2576 mice, reduces neuroinflammation caused by pathophysiological levels of plasma Ab, and reduces amyloid load and Ab40/42 levels in PD-hAbPP mice. (15a,33) Serum amyloid P (SAP) component can be removed from human amyloid deposits in peripheral tissues by drugs that are competitive inhibitors of SAP and may enable its rapid clearance (24) (see ARF related news story). It has been also suggested that insulin-like growth factor I may induce clearance of brain Ab, probably by enhancing the import into the CNS of Ab-carrier proteins such as albumin and transthyretin. (2) This therapeutic approach is alive and well, and we will discuss which gaps we must still fill in our basic understanding of Ab transport, which models are best suited to test the therapeutic approach, and whether current experimental compounds are promising.
Thumbnail Summary of Transport-Clearance Hypothesis
Out of the Brain: LRP-1 mediates rapid Ab transcytosis across the BBB. (29) In parallel, the interstitial fluid bulk flow into the CSF slowly removes soluble Ab. Into the Brain: RAGE mediates influx of free, circulating Ab across the BBB into the CNS. (11,15,16) Ab-sequestering agents such as sRAGE (15,33), anti-Ab IgG (3,30), gelsolin and/or GM1 (22), or sLRP-1 clusters II and IV (38) can mop up Ab in plasma, reducing its influx across the BBB. Eliminating contributions of the circulating pool of Ab to its central pool may promote Ab's flow from brain into blood; this could be particularly important in cases of sporadic AD where the efflux transport systems are defective, such as down-regulated LRP-1. (29)
We suggest these questions for discussion:
- What should the next experiments be to advance this potential therapeutic approach?
- Do receptor-mediated transport mechanisms respond in the same way to peripheral sequestration as passive flow would, i.e., will CNS levels go down if Ab is removed from the peripheral pool?
- Is serum Ab truly out of the picture once it is bound? Could transport systems "sense" that bound serum Ab is increasing, and reduce Ab efflux in response? If so, peripheral sequestration could lead to an unintended increase of CNS Ab. Have animal models ruled this out?
- Do peripheral Ab-antibody complexes get degraded faster or slower than Ab alone? Could they pile up and cause problems?
- The m266 work is a promising approach. How far along is it?
- Are there ways of stimulating peripheral degradation of Ab in the liver and the kidney?
- Does the importance of Ab clearance and peripheral degradation make AD a systemic disease?
- Is it desirable to solubilize aggregated Ab? Might soluble intermediates be toxic?
References:
1. Bading JR, Yamada S, Mackic JB, Kirkman L, Miller C, Calero M, Ghiso J, Frangione B, Zlokovic BV. Brain clearance of Alzheimer's amyloid-ß40 in the squirrel monkey: a SPECT study in a primate model of cerebral amyloid angiopathy. J Drug Targeting. 2002 10;4:359-368. Abstract
2. Carro E, Trejo JL, Gomez-Isla T, LeRoith D, Torres-Aleman I. Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nature Med. 2002;8:1390-1397. Abstract
3. DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-beta: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002;295:2264-2267. Abstract
4. DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-Ab antibody alters CNS and plasma Aß clearance and decreases brain Ab burden in a mouse model of lzheimer's disease. Proc Natl Acad Sci USA. 2001;98:8850-8855. Abstract
5. DeMattos RB, Bales KR, Parsadanian M, O'Dell MA, Foss EM, Paul SM, Holtzman DM. Plaque-associated disruption of CSF and plasma amyloid-beta equilibrium in a mouse model of Alzheimer's disease. J Neurochem. 2002;81:229-236. Abstract
6. DeMattos RB, Bales KR, Paul SM, Holtzman DM. Potential role of endogenous and exogenous Ab binding molecules in Ab clearance and metabolism. Ab Metabolism in Alzheimer's Disease. (Ed. T. Saido). Landes Bioscience. 2003;127-139.
7. Ertekin-Taner N, Graff-Radford N, Younkin LH, et al. Linkage of plasma Ab42 to a quantitative locus on chromosome 10 in late-onset Alzheimer's disease pedigrees. Science. 2000;290:2303-2304. Abstract
8. Ghersi-Egea JF, Gorevic PD, Ghiso J, Frangione B, Patlak CS, Fenstermacher JD. Fate of cerebrospinal fluid-borne amyloid b-peptide: rapid clearance into blood and appreciable accumulation by cerebral arteries. J Neurochem. 1996;67:880-83. Abstract
9. Ghilardi JR, Catton M, Stimson ER, Rogers S, Walker LC, Maggio JE, Mantyh PW. Intra arterial infusion of [125I]Ab1 40 labels amyloid deposits in the aged primate brain in vivo. Neuroreport. 1996;7:2607-2611. Abstract
10. Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest. 2001;108:779-784. Abstract
11. Hogg E, Kumar S, Holtzman J, et al. A model of brain arterial infusion in mice for the measurements of cerebrovascular functions: applications to amyloid b1-40 peptide transport in apolipoprotein E knockout mice. Soc Neurosci Abst. 2000;26:342.
12. Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee HJ, Hama E, Sekine-Aizawa Y, Saido TC. Y. Identification of the major Ab1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nature Med. 2000;6:143-150. Abstract
13. Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, Gerard C, Hama E, Lee HJ, Saido TC. Metabolic regulation of brain Ab by neprilysin. Science. 2001;292:1550-1552. Abstract
14. Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid b protein in the Tg2576 transgenic mouse model of Alzheimer's Disease. J Neuroscience. 2001;21:372-381. Abstract
15. Kumar R, Miao W, Ghiso J, et al. RAGE at the blood-brain barrier mediates neurovascular dysfunction caused by amyloid-b1-40 peptide. Soc Neuorsci Abst. 2000;26:741.
15A. Deane R, Yan SD, Kumar RS, LaRue B, Jovanovic S, Hogg E, Welch D, Maness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong D, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic BV. RAGE mediates amyloid-b peptide transport across the blood-brain barrier and accumulation in brain. Nature Medicine 2003, 9(7) in press. Advanced Online Publication June 15, 2003.
16. Mackic JB, Stins M, McComb JG, Calero M, Ghiso J, Kim KS, Yan SD, Stern D, Schmidt AM, Frangione B, Zlokovic BV. Human blood-brain barrier receptors for Alzheimer's amyloid-b. J Clin Invest. 1998;102:734-743. Abstract
17. Mackic JB, Weiss MH, Miao W, Kirkman E, Ghiso J, Calero M, Bading J, Frangione B, Zlokovic BV. Cerebrovascular accumulation and increased blood-brain barrier permeability to circulating Alzheimer's amyloid b-peptide in aged squirrel monkey with cerebral amyloid angiopathy. J. Neurochem. 1998;70:210-215. Abstract
18. Mackic JB, Bading J, Ghiso J, Walker L, Wisniewski T, Frangione B, Zlokovic BV. Differential cerebrovascular sequestration and enhanced blood-brain barrier permeability to circulating Alzheimer's amyloid-beta peptide in aged Rhesus vs. aged Squirrel monkey. Gen Pharmacology Vasc Biol. 2002;18:303-313.
19. Maness LM, Banks WA, Podlisny MB, Selkoe DJ, Kastin AJ. Passage of human amyloid-b protein 1-40 across the murine blood-brain barrier. Life Sci. 1994;55:1643-1650. Abstract
20. Martel CL, Mackic JB, McComb JG, Ghiso J, Zlokovic BV. Blood-brain barrier uptake of the 40 and 42 amino acid sequences of circulating Alzheimer's amyloid-b in guinea pigs. Neurosci Lett. 1996;206:157-160. Abstract
21. Martel CL, Mackic JB, Matsubara E, Governale S, Miguel C, Miao W, McComb JG, Frangione B, Ghiso J, Zlokovic BV. Isoform-specific effects of apolipoproteins E2, E3, E4 on cerebral capillary sequestration and blood-brain barrier transport of circulating Alzheimer's amyloid b. J Neurochem. 1997;69:1995-2004. Abstract
22. Matsuoka Y, Saito M, LaFrancois J, Saito M, Gaynor K, Olm V, Wang L, Casey E, Lu Y, Shiratori C, Lemere C, Duff K. Novel therapeutic approach for the treatment of Alzheimer's disease by peripheral administration of agents with an affinity to beta-amyloid. J Neurosci. 2003;23:29-33. Abstract (See also ARF related news story)
23. Monro OR, Mackic JB, Yamada S, Segal MB, Ghiso J, Maurer C, Calero M, Frangione B, Zlokovic BV. Substitution at codon 22 reduces clearance of Alzheimer's amyloid-b peptide from the cerebrospinal fluid and prevents its transport from the central nervous system into blood. Neurobiol Aging. 2002;23:405-412. Abstract
24. Pepys MB, Herbert J, Hutchinson GA, et al. Targeted pharmacological deletion of serum amyloid P component for treatment of human amyloidosis. Nature. 2002;417:254-259. Abstract
25. Poduslo JF, Curran GL, Haggard JJ, Biere AL, Selkoe DJ. Permeability and residual plasma volume of human, Dutch variant, and rat amyloid b-protein 1-40 at the blood-brain barrier. Neurobiol Dis. 1997;4:27-34. Abstract
26. Poduslo JF, Curran GL. Amyloid-b peptide as a vaccine for Alzheimer's disease involves receptor-mediated transport at the blood-brain barrier. Neuroreport. 2001;12:3197-3200. Abstract
27. Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol. 1998;8:447-453. Abstract
28. Selkoe DJ. Clearing the brain's amyloid cobwebs. Neuron. 2001;32:177-180. Abstract
29. Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer's amyloid-b1- 40 peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489-1499. Abstract
30. Sigurdsson EM, Scholtzova H, Mehta PD, et al. Immunization with a nontoxic/nonfibrillar amyloid-beta homologous peptide reduces Alzheimer's disease-associated pathology in transgenic mice. Am J Pathol. 2001;159:439-447. Abstract
31. Stern D, Yan SD, Yan SF, et al. Receptor for advanced glycation endproducts: a multiligand receptor magnifying cell stress in diverse pathologic settings. Adv Drug Del Rev. 2002;54:1615-1625. Abstract
32. Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685-691. Abstract
33. Yu J, Zhu H, Pettigrew LC, et al. Infusion of soluble RAGE inhibits Ab amyloid deposition in AbPP transgenic mice. Soc Neurosci Abstr. 2001;27:856.
34. Zlokovic BV, Ghiso J, Mackic JB, McComb JG, Weiss MH, Frangione B. Blood-brain barrier transport of circulating Alzheimer's amyloid-beta. Biochem Biophys Res Commun. 1993;197:1034-40. Abstract
35. Zlokovic BV, Martel CL, Matsubara E, McComb JG, Zheng G, McCluskey RT, Frangione B, Ghiso J. Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer's disease amyloid-b ?at the blood-brain and blood-cerebrospinal fluid barriers. Proc Natl Acad Sci USA. 1996;93:4229-4234. Abstract
36. Zlokovic BV, Yamada S, Holtzman D, Ghiso J, Frangione B. Clearance of amyloid-beta peptide from brain: transport or metabolism? Nature Med. 2000;6:718-719. Abstract
37. Zlokovic BV, Frangione B. Transport-clearance hypothesis for Alzheimer's disease and potential therapeutic implications. Ab Metabolism in Alzheimer's Disease. (Ed. T. Saido). Landes Bioscience. 2003;114-122.
38. Zlokovic BV, Wu Z, Barclay DR, Lenting PJ, Yan S, Deane R, Pinkert CA. LRP-1 binds free Ab and promotes its clearance across the blood-brain barrier in wild type and Tie-2 LRP-1 Tg mice. Soc. Neurosci. 2003 (in press).
References
News Citations
- Early Diagnosis of Alzheimer's—Making Use of the Blood-Brain Barrier
- Zapping Amyloid with SAP Inhibitors
- Budding RNAi Therapies, APP Protein Interaction Map Impress at Meeting
- Got Plaques? Astrocytes to the Rescue!
Webinar Citations
Paper Citations
- Bading JR, Yamada S, Mackic JB, Kirkman L, Miller C, Calero M, Ghiso J, Frangione B, Zlokovic BV. Brain clearance of Alzheimer's amyloid-beta40 in the squirrel monkey: a SPECT study in a primate model of cerebral amyloid angiopathy. J Drug Target. 2002 Jun;10(4):359-68. PubMed.
- Carro E, Trejo JL, Gomez-Isla T, LeRoith D, Torres-Aleman I. Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat Med. 2002 Dec;8(12):1390-7. PubMed.
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001 Jul 17;98(15):8850-5. Epub 2001 Jul 3 PubMed.
- Demattos RB, Bales KR, Parsadanian M, O'Dell MA, Foss EM, Paul SM, Holtzman DM. Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer's disease. J Neurochem. 2002 Apr;81(2):229-36. PubMed.
- Ertekin-Taner N, Graff-Radford N, Younkin LH, Eckman C, Baker M, Adamson J, Ronald J, Blangero J, Hutton M, Younkin SG. Linkage of plasma Abeta42 to a quantitative locus on chromosome 10 in late-onset Alzheimer's disease pedigrees. Science. 2000 Dec 22;290(5500):2303-4. PubMed.
- Ghersi-Egea JF, Gorevic PD, Ghiso J, Frangione B, Patlak CS, Fenstermacher JD. Fate of cerebrospinal fluid-borne amyloid beta-peptide: rapid clearance into blood and appreciable accumulation by cerebral arteries. J Neurochem. 1996 Aug;67(2):880-3. PubMed.
- Ghilardi JR, Catton M, Stimson ER, Rogers S, Walker LC, Maggio JE, Mantyh PW. Intra-arterial infusion of [125I]A beta 1-40 labels amyloid deposits in the aged primate brain in vivo. Neuroreport. 1996 Nov 4;7(15-17):2607-11. PubMed.
- Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest. 2001 Sep;108(6):779-84. PubMed.
- Iwata N. [Identification of the major A beta 1-42-degrading catabolic pathway in brain parenchyma]. Seikagaku. 2003 Feb;75(2):97-107. PubMed.
- Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, Gerard C, Hama E, Lee HJ, Saido TC. Metabolic regulation of brain Abeta by neprilysin. Science. 2001 May 25;292(5521):1550-2. PubMed.
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001 Jan 15;21(2):372-81. PubMed.
- Mackic JB, Stins M, McComb JG, Calero M, Ghiso J, Kim KS, Yan SD, Stern D, Schmidt AM, Frangione B, Zlokovic BV. Human blood-brain barrier receptors for Alzheimer's amyloid-beta 1- 40. Asymmetrical binding, endocytosis, and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J Clin Invest. 1998 Aug 15;102(4):734-43. PubMed.
- Mackic JB, Weiss MH, Miao W, Kirkman E, Ghiso J, Calero M, Bading J, Frangione B, Zlokovic BV. Cerebrovascular accumulation and increased blood-brain barrier permeability to circulating Alzheimer's amyloid beta peptide in aged squirrel monkey with cerebral amyloid angiopathy. J Neurochem. 1998 Jan;70(1):210-5. PubMed.
- Maness LM, Banks WA, Podlisny MB, Selkoe DJ, Kastin AJ. Passage of human amyloid beta-protein 1-40 across the murine blood-brain barrier. Life Sci. 1994;55(21):1643-50. PubMed.
- Martel CL, Mackic JB, McComb JG, Ghiso J, Zlokovic BV. Blood-brain barrier uptake of the 40 and 42 amino acid sequences of circulating Alzheimer's amyloid beta in guinea pigs. Neurosci Lett. 1996 Mar 15;206(2-3):157-60. PubMed.
- Martel CL, Mackic JB, Matsubara E, Governale S, Miguel C, Miao W, McComb JG, Frangione B, Ghiso J, Zlokovic BV. Isoform-specific effects of apolipoproteins E2, E3, and E4 on cerebral capillary sequestration and blood-brain barrier transport of circulating Alzheimer's amyloid beta. J Neurochem. 1997 Nov;69(5):1995-2004. PubMed.
- Matsuoka Y, Saito M, LaFrancois J, Gaynor K, Olm V, Wang L, Casey E, Lu Y, Shiratori C, Lemere C, Duff K. Novel therapeutic approach for the treatment of Alzheimer's disease by peripheral administration of agents with an affinity to beta-amyloid. J Neurosci. 2003 Jan 1;23(1):29-33. PubMed.
- Monro OR, Mackic JB, Yamada S, Segal MB, Ghiso J, Maurer C, Calero M, Frangione B, Zlokovic BV. Substitution at codon 22 reduces clearance of Alzheimer's amyloid-beta peptide from the cerebrospinal fluid and prevents its transport from the central nervous system into blood. Neurobiol Aging. 2002;23(3):405-12. PubMed.
- Pepys MB, Herbert J, Hutchinson WL, Tennent GA, Lachmann HJ, Gallimore JR, Lovat LB, Bartfai T, Alanine A, Hertel C, Hoffmann T, Jakob-Roetne R, Norcross RD, Kemp JA, Yamamura K, Suzuki M, Taylor GW, Murray S, Thompson D, Purvis A, Kolstoe S, Wood SP, Hawkins PN. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature. 2002 May 16;417(6886):254-9. PubMed.
- Poduslo JF, Curran GL, Haggard JJ, Biere AL, Selkoe DJ. Permeability and residual plasma volume of human, Dutch variant, and rat amyloid beta-protein 1-40 at the blood-brain barrier. Neurobiol Dis. 1997;4(1):27-34. PubMed.
- Poduslo JF, Curran GL. Amyloid beta peptide as a vaccine for Alzheimer's disease involves receptor-mediated transport at the blood-brain barrier. Neuroreport. 2001 Oct 29;12(15):3197-200. PubMed.
- Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol. 1998 Nov;8(11):447-53. PubMed.
- Selkoe DJ. Clearing the brain's amyloid cobwebs. Neuron. 2001 Oct 25;32(2):177-80. PubMed.
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer's amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000 Dec;106(12):1489-99. PubMed.
- Sigurdsson EM, Scholtzova H, Mehta PD, Frangione B, Wisniewski T. Immunization with a nontoxic/nonfibrillar amyloid-beta homologous peptide reduces Alzheimer's disease-associated pathology in transgenic mice. Am J Pathol. 2001 Aug;159(2):439-47. PubMed.
- Stern D, Yan SD, Yan SF, Schmidt AM. Receptor for advanced glycation endproducts: a multiligand receptor magnifying cell stress in diverse pathologic settings. Adv Drug Deliv Rev. 2002 Dec 7;54(12):1615-25. PubMed.
- Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996 Aug 22;382(6593):685-91. PubMed.
- Zlokovic BV, Ghiso J, Mackic JB, McComb JG, Weiss MH, Frangione B. Blood-brain barrier transport of circulating Alzheimer's amyloid beta. Biochem Biophys Res Commun. 1993 Dec 30;197(3):1034-40. PubMed.
- Zlokovic BV, Martel CL, Matsubara E, McComb JG, Zheng G, McCluskey RT, Frangione B, Ghiso J. Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood-brain and blood-cerebrospinal fluid barriers. Proc Natl Acad Sci U S A. 1996 Apr 30;93(9):4229-34. PubMed.
- Zlokovic BV, Yamada S, Holtzman D, Ghiso J, Frangione B. Clearance of amyloid beta-peptide from brain: transport or metabolism?. Nat Med. 2000 Jul;6(7):718-9. PubMed.
- Koudinov AR, Koudinova NV. Alzheimer's soluble amyloid beta protein is secreted by HepG2 cells as an apolipoprotein. Cell Biol Int. 1997 May;21(5):265-71. PubMed.
Other Citations
Further Reading
No Available Further Reading
Panelists
-
 Berislav Zlokovic, M.D., Ph.D.
University of Southern California
Berislav Zlokovic, M.D., Ph.D.
University of Southern California
Comments
Emory University
It appears likely that the abnormal conformation and assembly of specific proteins underlies a surprising variety of degenerative diseases of the brain and other organs. How proteins such as Ab cause neurodegeneration in Alzheimer's disease is still a matter of dispute. Although autosomal-dominant cases of AD are relatively rare, it is telling that every known genetic risk factor for AD increases the production and or autophilicity of Ab, resulting in a buildup of the protein in brain. Because many cases of AD have no obvious genetic link, other mechanisms that promote the focal accumulation of Ab must be considered.
Given that the concentration of specific proteins is a key determinant of the likelihood that they will form aggregates (Lansbury, 1997), the therapeutic options for impeding the proteopathic cascade boil down to one theme: Prevent the proteins from achieving critical mass for aggregation into oligomeric and/or fibrillar forms. This can be done by reducing the genesis of the offending proteins (e.g., via secretase inhibitors), blocking their self-assembly (e.g., with aggregation inhibitors), or promoting their dispersal (e.g., by creation of a peripheral "sink"). The Zlokovic group (Deane et al., 2003) presents nice data indicating that circulating, free Ab is ferried into brain by the endothelial receptor for advanced glycation end products (RAGE). Blocking this transport (e.g., by binding circulating Ab with soluble RAGE) diminishes Ab load in AbPP-transgenic mouse brain, presumably by favoring efflux of the protein from brain parenchyma. This group also has found in nonhuman primates that cerebral Ab deposition correlates with an age-related decline in the clearance of Ab from brain (Bading et al., 2002; Mackic et al., 2002). Overall, the results support the idea that altering the equilibrium of Ab in favor of dispersal from brain can lower the odds of aberrant aggregation and, possibly, the emergence of clinical disease.
An intriguing transgenic model was introduced a few years back in which carboxyl-terminal AbPP is expressed in mice via a cytomegalovirus enhancer/chick b-actin promoter (Fukuchi et al., 1996). Transgene expression is found in many tissues, but it is highest in heart, muscle, and intestine, and comparatively low in brain. Despite high levels of circulating human Ab (17 times normal human plasma levels), the mice fail to develop cerebral b-amyloidosis, even up to 29 months of age. (Interestingly, the mice do show Ab deposits in other organs, such as intestine and muscle; see Fukuchi et al., 1996 and Fukuchi et al., 1998.) It would be informative, in light of the peripheral sink hypothesis, to know why senile plaques and cerebrovascular b-amyloidosis are absent in these mice.
Finally, there is in-vitro evidence that the transporter p-glycoprotein (P-gp) acts as a cellular efflux pump for Ab (Lam et al., 2001). The function of P-gp in the brain is poorly understood, but it is known to be particularly abundant in the vascular endothelium (Golden and Pardridge, 2000). Interestingly, the vascular expression of P-gp correlates inversely with brain Ab load in aged humans (Vogelgesang et al., 2002), suggesting that this transporter also may contribute to the regulation of Ab levels in brain, and could theoretically compensate for changes in the activity of other transport mechanisms. Although the regulation of the brain chemical milieu is complex, modulation of protein transport is an exciting new option for the possible treatment of AD and other proteopathies.
References:
Lansbury PT. Structural neurology: are seeds at the root of neuronal degeneration?. Neuron. 1997 Dec;19(6):1151-4. PubMed.
Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003 Jul;9(7):907-13. PubMed.
Bading JR, Yamada S, Mackic JB, Kirkman L, Miller C, Calero M, Ghiso J, Frangione B, Zlokovic BV. Brain clearance of Alzheimer's amyloid-beta40 in the squirrel monkey: a SPECT study in a primate model of cerebral amyloid angiopathy. J Drug Target. 2002 Jun;10(4):359-68. PubMed.
Mackic JB, Bading J, Ghiso J, Walker L, Wisniewski T, Frangione B, Zlokovic BV. Circulating amyloid-beta peptide crosses the blood-brain barrier in aged monkeys and contributes to Alzheimer's disease lesions. Vascul Pharmacol. 2002 Jun;38(6):303-13. PubMed.
Fukuchi K, Ho L, Younkin SG, Kunkel DD, Ogburn CE, Leboeuf RC, Furlong CE, Deeb SS, Nochlin D, Wegiel J, Wisniewski HM, Martin GM. High levels of circulating beta-amyloid peptide do not cause cerebral beta-amyloidosis in transgenic mice. Am J Pathol. 1996 Jul;149(1):219-27. PubMed.
Fukuchi K, Pham D, Hart M, Li L, Lindsey JR. Amyloid-beta deposition in skeletal muscle of transgenic mice: possible model of inclusion body myopathy. Am J Pathol. 1998 Dec;153(6):1687-93. PubMed.
Lam FC, Liu R, Lu P, Shapiro AB, Renoir JM, Sharom FJ, Reiner PB. beta-Amyloid efflux mediated by p-glycoprotein. J Neurochem. 2001 Feb;76(4):1121-8. PubMed.
Golden PL, Pardridge WM. Brain microvascular P-glycoprotein and a revised model of multidrug resistance in brain. Cell Mol Neurobiol. 2000 Apr;20(2):165-81. PubMed.
Vogelgesang S, Cascorbi I, Schroeder E, Pahnke J, Kroemer HK, Siegmund W, Kunert-Keil C, Walker LC, Warzok RW. Deposition of Alzheimer's beta-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics. 2002 Oct;12(7):535-41. PubMed.
Make a Comment
To make a comment you must login or register.