Past Webinar
Can Network Analysis Identify Pathological Pathways in Alzheimer’s
Quick Links
Introduction
In the April 25 Cell, Valur Emilsson at the Icelandic Heart Association and Eric Schadt at Icahn School of Medicine at Mount Sinai, New York, report that they have identified molecular networks that are perturbed in Alzheimer’s disease patients compared to normal, age-matched controls. Several of these networks comprise genes previously linked to AD, including TREM2 and CD33. The scientists also identified a new player, TYROBP, as a master regulator of these molecular modules. Meanwhile, in the April 25 Neuron, researchers led by Rudy Tanzi and Ana Griciuc at Massachusetts General Hospital, Charlestown, report that microglia in the AD brain overproduce CD33, which seems to prevent these cells from binding to and degrading amyloid-β. Together, these findings tighten the link between AD pathology and microglial dysfunction.
Alzforum held a Webinar on Thursday, 23 May 2013, with Eric Schadt and Ana Griciuc, along with Monica Carson, University of California, Riverside; John Hardy, University College London; and Jeremy Miller, Allen Institute for Brain Science, Seattle, Washington.
We thank Cell Press for giving Alzforum readers temporary free access to these papers:
 Zhang B, Gaiteri C, Bodea L-G, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Suver C, Shah H, Mahajan M, Gillis T, Mysore J, MacDonald ME, Lamb JR, Bennett DA, Molony C, Stone DJ, Gudnason V, Myers AJ, Schadt EE, Neumann H, Zhu J, Emilsson V. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013 April 25 2013;153:707-720. Read article.
Zhang B, Gaiteri C, Bodea L-G, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Suver C, Shah H, Mahajan M, Gillis T, Mysore J, MacDonald ME, Lamb JR, Bennett DA, Molony C, Stone DJ, Gudnason V, Myers AJ, Schadt EE, Neumann H, Zhu J, Emilsson V. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013 April 25 2013;153:707-720. Read article.
 Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid β. Neuron. 2013 April 25 online. Read article.
Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid β. Neuron. 2013 April 25 online. Read article.
Gandy S, Heppner FL. Microglia as Dynamic and Essential Components of the Amyloid Hypothesis. Neuron. 2013 May 22. Read article.
- Eric Schadt's Presentation
- Ana Griciuc's Presentation
- Monica Carson's Presentation
- John Hardy's Presentation
- Jeremy Miller's Presentation
Background
Background Text
By Tom Fagan
Last year, two groups uncovered mutations in the gene for TREM2 that multiply a person’s risk for Alzheimer’s disease about threefold, making TREM2 the second most important genetic risk factor after ApoE (see ARF related news story). How does TREM2 influence pathogenesis? It encodes a membrane receptor found on different cell types. It may interact with many molecular partners, potentially influencing an untold number of different cellular pathways. The same goes for CD33, an immunoglobulin-like cell-surface receptor of unknown function. While researchers are encouraged that the genetic data point to specific cells or systems, such as microglia or innate immunity, the exact molecular pathways involved in pathogenesis have yet to be worked out. As for the other AD risk genes that emerged from genomewide association studies, including Bin1, CLU, PICALM, CD33, MS4A6A, and MS4A6E (see Alzgene Top Results), some fall into those pathways, but scientists are unsure about their exact role in AD.
How can scientists match genetic variation to function? One approach would be to painstakingly investigate each gene's so-called interactome—that is, look at binding partners and associated pathways and test them for pathogenicity. Alternatively, scientists could correlate known genetic risk factors with processes that are known to have gone awry. Here, systems biology can help. In recent years, researchers have used whole-genome expression data to identify groups of genes, aka transcription modules, that are coordinately expressed and functionally related (see Zhang and Horvath, 2005). This type of network analysis has the potential to uncover, in one experiment, whole pathways that may be perturbed in disease.
Schadt and colleagues have used this approach to identify molecular modules that might contribute to metabolic syndrome (Chen et al., 2008), and others have studied hippocampal networks in Alzheimer's disease similarly (see ARF related news story on Miller et al., 2008). More specifically, this approach can relate genetic hits to biological pathways. For example, researchers have used network analysis to link progranulin mutations to the Wnt signaling pathway in frontotemporal dementia (see ARF related news story).
Now, Schadt and collaborators take a systems approach to uncover dysfunctional expression throughout the whole AD brain. They used network analysis, comparing gene expression patterns in tissue taken from patients and controls, to identify molecular modules that are reconfigured in anatomical regions most devastated in late-onset AD. Network disturbances related to immunity and microglia topped the list. In particular, they predict that the microglial protein TYROBP, a TREM2 binding partner, acts as a master regulator of an innate immunity module that includes other AD GWAS hits MS4A6A, MS4A6E, and CD33. Hematopoietic cells, including neutrophils, macrophages, natural killer cells, and B and T cells make TYROBP. The AD brain seems to overproduce the protein, the scientists report.
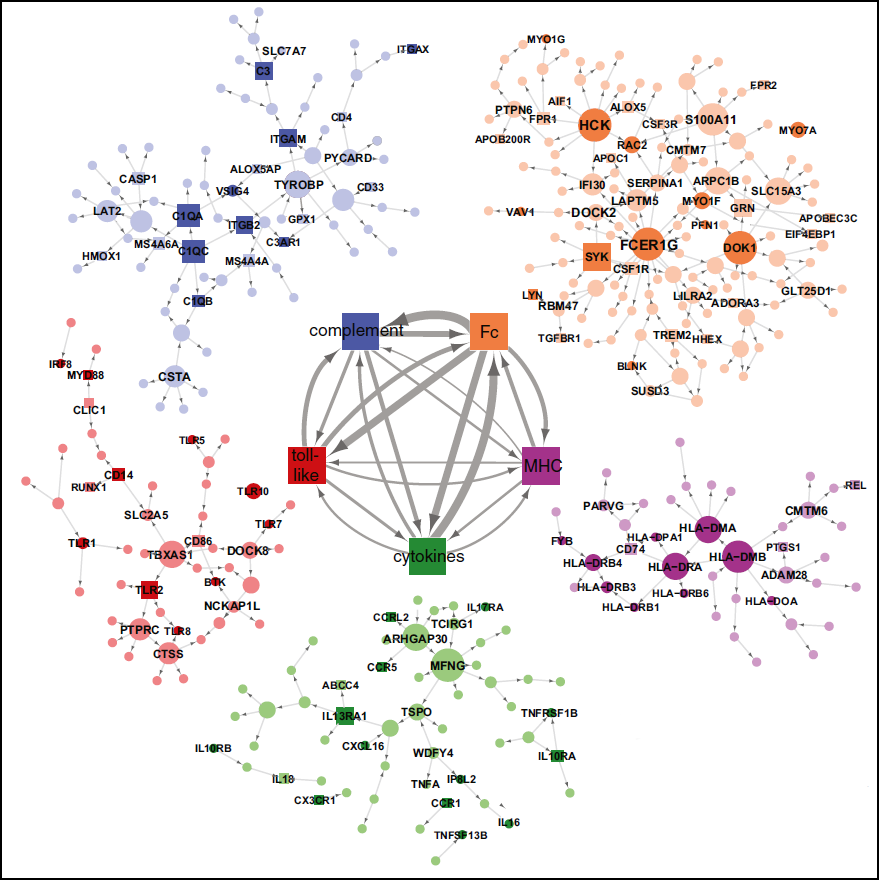
AD-specific molecular networks. Image courtesy of Cell Press
How might TYROBP knock molecular networks off balance in AD? A hint comes from Ana Griciuc and Rudy Tanzi. They found elevated expression of CD33 in microglial cells in AD brain samples, and report that overexpression of this protein by microglia in culture impairs their ability to take up and degrade amyloid-β. Consistent with this, they found that in AD brain samples, greater numbers of CD33-positive microglia correlated with higher amyloid burden. Interestingly, carriers of the protective CD33 genetic variant had less of this immunoglobulin in the brain, suggesting that reducing the protein might prove beneficial. In fact, knocking out CD33 in APP/PS1 transgenic mice dramatically reduced amyloid plaques. These findings strengthen the case that TYROBP-regulated pathways, and microglial dysfunction in general, play a role in sporadic AD pathology.
Q: It would be interesting to see how these microglial networks evolve during normal human aging, and whether they are shaped differently in young versus old people.
Miller: This would be interesting and potentially could be studied using the data from Eric's paper. This is also being studied in mice in the lab of Erik Boddeke at the University of Groningen, the Netherlands. They have a technique for purifying microglia, and are doing so from several models of accelerated aging (as well as normal aging). One of the things they are looking at is how gene networks change within these pure cell populations. This analysis is ongoing. I do not know of any labs specifically addressing this issue in humans.
Schadt: Age is one of the most significant covariates for both AD and non-demented expression data. Several of the subnetworks are enriched for genes associated with aging, so it would absolutely be interesting to more strongly leverage the data we published to examine aging induced changes in microglial cells.
Q: The AD brain exhibits marked changes in the numbers of neurons and glial cells relative to the non-demented brain. There is a strong depletion of neurons and a dramatic increase in the number of microglial cells and astrocytes. Have you corrected/normalized for the abundance of neurons/glial cells? Is the TYROBP-related microglial network so prominent because the AD brain has a dramatic enrichment in microglia?
Miller: In general, network analysis based on samples of heterogeneous tissue is very good at identifying groups of coexpressed genes corresponding to different cell types. Therefore, it would not surprise me if the prominence of the microglial module as a whole was due to the changes in relative numbers of neurons and glia in AD. However, the differential connectivity of genes within this module should be much less related to the number of microglia, and much more related to how these cells change in AD compared with controls.
Schadt: Changes in the composition of the cell types of the brain regions profiled will certainly influence the absolute levels of expression of genes detected. Given that genes expressed in one cell type but not in others (or differentially expressed in other cell types) will be detected as an average overall cell type, variations in cell-type composition will affect observed expression levels. However, expression changes would be linear if only due to changes in cell-type composition, and since the correlations we computed among expression traits are invariant under linear translations, cell-type shifts would not be expected to influence the correlation structure depicted in our networks. However, if the shift in cell composition was the result of, or resulted in, functional changes, then the correlations would be affected, but this would then not be an artifact of changes in cell-type composition but rather would convey meaningful functional changes.
I had also noted that, surprisingly, there were only a couple of hundred genes that were differentially expressed between AD and non-demented brains, so that even though we had high power to detect differential expression, we detected an order of magnitude or more fewer differentially expressed genes than differentially connected genes, supporting that changes in cell-type composition had likely modest, at best, impact on absolute expression (again we would expect changes in cell-type composition to most profoundly affect differential expression measures, not differential connectivity). In addition, the fact that we leveraged eSNPs as a perturbation source to inform the networks minimizes the impact of changes in expression driven by changes in cell-type composition. Germline DNA is not changing (certainly not at the same rate) like RNA changes, and it is this fixed nature of DNA and its association to expression levels that enable us to tease apart the genetic variance component in the expression data from the environmental variance component.
Q: How disease specific are these networks?
Schadt: The networks we identified as strongly associated with AD are definitely not disease specific, but instead we believe they reflect how changes in macrophage-like function can predispose to disease onset and/or severity. We have implicated the network we identified for AD in a range of metabolic and autoimmune disorders, such as obesity, diabetes, heart disease, and irritable bowel disease (IBD). It would seem that dysfunction in this network, say, in fat, predisposes to diabetes; in gut, predisposes to IBD; in brain, predisposes to AD; and so on (see Wang et al., 2012, for a list of diseases we have linked to this network).
Q: When analyzing gene expression and proteomics in AD brain tissue, I would expect neurodegeneration and cell death, including apoptosis, to severely affect and dominate the results. How do you handle that when interpreting data?
Miller: This can be done in a few ways. First, one can correct for it using a linear model with well-established markers for key cell types. Second, one could identify modules likely to correspond to different cell types and then see how the genes in these modules change relative to one another in control versus disease—this is the idea behind differential connectivity. Conversely, it is also possible to use these literature- or network-derived genes corresponding to neurons and glia to estimate the amount of neurodegeneration and gliosis in each sample. This would obviously not be as accurate as experimental validation, but can give a reasonable estimate.
Schadt: In addition to the above ways, the genetic dimension, again, is very good for teasing apart the genetic and environmental variance components for the molecular trait data, so that the detection of eQTL (eSNPs) and the impacts these have on expression that associates with AD, provides a very clean path to focusing on direct causes as opposed to effects. As indicated above, germline DNA, as we use it, would not be changing between AD and controls.
Q: Postmortem tissue is obviously not similar to living tissue. How is this taken into account?
Miller: There are various techniques one can use to make sure that individual samples are not bad. These "outlier" samples can be removed from the experiment or the analysis depending on when the bad samples are identified. While it is not possible to prove that the samples completely recapitulate living tissue, it is very possible to make sure that all of the postmortem samples have relatively similar transcription to one another and should therefore be comparable to one another.
Schadt: Again, the DNA dimension here helps as well, since the germline DNA as we would use it in this study was not subject to change given the postmortem status. So the variations in “fixed” DNA that associate with expression levels I think serves as a pretty good filter for those processes that would be changing after death. In addition, the correlation of these traits to other clinical features such as AD status, pathophysiology, sex, age, etc., would also help tune you into the right signal. Getting postmortem samples is obviously not ideal, but that is, today, the only way we can get such samples. We are also actively generating iPSCs from AD and control patients and transforming them into mixtures of cell types that include glial cells. That will provide another way to assess what parts of the network from postmortem tissue may be most relevant to disease causation and progression.
Q: What is the quality of human brain autopsy tissue needed to do the expression studies? How long can the tissue be frozen to get good data?
Miller: In my experience, there is no set cutoff time, although it is always good to limit the number of freeze-thaw cycles of any given tissue.
Schadt: We have found that despite some of the brain samples having been on ice for 48 or more hours, many times the quality of the RNA is adequate to get the type of results we reported. That is pretty amazing, in my opinion, and it is unlike what we see in other tissues such as liver and fat, where long periods on ice renders them almost completely useless for these purposes. It may be that the blood-brain barrier better protects the brain from the rapid degradation as we find in other tissues. It is very difficult to collect brain tissue that can be flash frozen at the time of death, so fortunately, the RNA is relatively stable, at least in the parts we profiled (DNA is far more stable and there is no variation in quality, from the standpoint of genotype cell rate, as a function of the time the brain sat on ice).
Q: How do the network pathways identified and the microglia link to tau tangles?
Miller: I have noticed that some of the same microglial genes discussed in the Webinar also show increased expression in tangle-bearing neurons compared with nearby non-tangle-bearing neurons from the entorhinal cortex of AD patients. This suggests that microglia show an increased presence around tangle-bearing neurons, although it is not possible to say whether the microglia or the tangles are there first based on these data alone. The original study discussing this is freely available (see Dunckley at al., 2006).
Schadt: Others have associated gene expression traits in the network we identified in AD with tau tangles induced in mice by a high-fat athero diet (see Krass et al., 2003).
Q: Does it make any difference to have TREM2 as a hub, or to have TYROBP as a hub in this network?
Hardy: No, they are close to each other.
Q: What effect does upregulation of TREM2 have on the ongoing proinflammatory phenotype? What I understood from the Webinar is that TREM1 signaling is more proinflammatory and TREM2 signaling more suppressive?
Carson: This is an interesting issue, especially since both molecules use DAP12 (also called TYROBP) as their transmembrane signaling partner. Transfection of TREM1-expressing cells with TREM2 vectors does decrease the production of several proinflammatory cytokines. However, the original publications by Colonna and colleagues indicated that TREM2 stimulated dendritic cell upregulation of CCR7, MHC Class II, B7.2 (CD86) and CD40—molecules usually associated with classical activation (see Bouchon et al., 2001). In microglia, TREM2 also promotes phagocytic activity and induction of key molecules required for antigen presentation. Knockdown and overexpression studies confirmed that TREM2hi microglial cell lines were better at stimulating T cell proliferation and cytokine production than TREM2lo microglial cell lines (see Melchior et al., 2010). Thus, while TREM2 is clearly immunomodulatory, it may be an oversimplification to consider it strictly as anti-inflammatory.
Q: Can you clarify that TYROBP is activated in AD, yet loss of function of TREM2 is associated with AD?
Carson: In the standard mouse models of amyloid pathology, both TYROBP (also called DAP12) and TREM2 are upregulated in the same set of microglia immediately surrounding the amyloid plaques (Figs. in Melchior et al., 2010). TYROBP expression is required for TREM2 to reach the cell surface. TREM2 lacks an intracellular signaling tail and requires TYROBP to mediate its intracellular signaling. Homozygous loss of TREM2 function in humans does not result in AD; it results in an early-onset cognitive dementia called Nasu-Hakola disease (apparent much earlier, in the twenties) that is distinct from classic AD. Loss of TYROBP results in the same Nasu-Hakola disease. Thus, the newly reported mutations in TREM2 are likely not complete loss of function.
Hardy: We have looked at this and not found evidence for it, but we will look again when we have a larger sample set.
Q: Since inflammation is a double-edged sword, having both neuroprotective and neurotoxic effects, do you think that it will be a good therapeutic approach to inhibit microglia activation, or should there be an effort to investigate which factors are toxic and which are protective?
Carson: This is a topic of high debate. My opinion is that microglia have multiple homeostatic functions in the healthy brain and can acquire multiple different types of effector states as part of an inflammatory response. Therefore, global suppression of all their functions will likely have detrimental effects. In addition, the definition of toxic versus protective responses may be context specific and change as the brain ages. The challenge is developing therapies that selectively target maladaptive versus adaptive pathways at each age and disease stage.
Q: You mentioned alternative routes to immune regulation via interleukin-1β receptor antagonists when we discussed TREM2 as a target. Can you expand on that thinking or clarify?
Carson: I apologize for any confusion. To clarify, I would suggest we can learn from examples provided by the IL-1β system. IL-1β signals through IL-1 receptor 1. However, IL-1 receptor 2 acts as an antagonist in part because it is shed and acts as a decoy receptor (binds IL-1β so that IL-1β cannot bind IL-1 receptor 1).
TREM2 exists as a membrane-bound form that can pair with TYROBP (DAP12) to become a signaling complex. However, TREM2 also exists as a non-membrane-bound form. This form has the potential to be a decoy receptor as well, preventing TREM2 ligands from binding the membrane-bound form of TREM2. Thus, when we consider the consequences of increased transcription of TREM2, the consequences may be due to the "decoy," the membrane-bound form, or both!
Q: Have you characterized individuals that carry the ApoE4 risk allele and TREM2 risk alleles? Do they have a synergistic enhancement of brain pathology and a further decrease in cognitive performance? Can human genetics offer insights into potential cooperations between ApoE and other risk genes in regulating AD pathology?
Hardy: Yes, we have looked at this, but the sample size was too small to make any conclusions.
Q: How do differences in mouse/human CD33 signaling affect interpretation of your results, given that mouse CD33 is quite different from human CD33, with a different sugar ligand and an opposite signaling pathway. Human CD33 signals inhibitory via immunoreceptor tyrosine-based activation motif (ITIM). Mouse CD33 has a charged residue binding to TYROBP—possible immunoreceptor tyrosine-based activation motif (ITAM) signaling.
Griciuc: Our experiments using primary microglial cultures derived from CD33-null mice, as well as our phenotypic analysis of APP/PS1 mice in a CD33-null background, indicate that mouse CD33 modulates amyloid-β clearance the same way human CD33 does. Mouse CD33 contains a charged residue in the transmembrane region and an ITIM-like domain in the cytosolic region; however, there is no published work showing that mouse CD33 recruits TYROBP and performs ITAM signaling. The presence of the inhibitory ITIM-like domain in mouse CD33 would rather suggest that it performs inhibitory signaling, similar to human CD33. Our current experiments are addressing the signaling pathways activated by mouse and human CD33. We are also addressing whether the presence of amyloid-β alters the CD33-mediated signaling in microglial cells, and are also comparing the pathways activated by several other substrates.
References
News Citations
- Enter the New Alzheimer’s Gene: TREM2 Variant Triples Risk
- Required Reading—InteracTomes for AD, Aging, APP
- Systems Biology Approaches Get Wnt of Progranulin’s Role in FTD
Paper Citations
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. PubMed.
- Chen Y, Zhu J, Lum PY, Yang X, Pinto S, Macneil DJ, Zhang C, Lamb J, Edwards S, Sieberts SK, Leonardson A, Castellini LW, Wang S, Champy MF, Zhang B, Emilsson V, Doss S, Ghazalpour A, Horvath S, Drake TA, Lusis AJ, Schadt EE. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008 Mar 27;452(7186):429-35. PubMed.
- Miller JA, Oldham MC, Geschwind DH. A systems level analysis of transcriptional changes in Alzheimer's disease and normal aging. J Neurosci. 2008 Feb 6;28(6):1410-20. PubMed.
- Wang IM, Zhang B, Yang X, Zhu J, Stepaniants S, Zhang C, Meng Q, Peters M, He Y, Ni C, Slipetz D, Crackower MA, Houshyar H, Tan CM, Asante-Appiah E, O'Neill G, Luo MJ, Thieringer R, Yuan J, Chiu CS, Lum PY, Lamb J, Boie Y, Wilkinson HA, Schadt EE, Dai H, Roberts C. Systems analysis of eleven rodent disease models reveals an inflammatome signature and key drivers. Mol Syst Biol. 2012;8:594. PubMed.
- Dunckley T, Beach TG, Ramsey KE, Grover A, Mastroeni D, Walker DG, LaFleur BJ, Coon KD, Brown KM, Caselli R, Kukull W, Higdon R, McKeel D, Morris JC, Hulette C, Schmechel D, Reiman EM, Rogers J, Stephan DA. Gene expression correlates of neurofibrillary tangles in Alzheimer's disease. Neurobiol Aging. 2006 Oct;27(10):1359-71. PubMed.
- Krass KL, Colinayo V, Ghazalpour A, Vinters HV, Lusis AJ, Drake TA. Genetic loci contributing to age-related hippocampal lesions in mice. Neurobiol Dis. 2003 Jul;13(2):102-8. PubMed.
- Bouchon A, Hernández-Munain C, Cella M, Colonna M. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J Exp Med. 2001 Oct 15;194(8):1111-22. PubMed.
- Melchior B, Garcia AE, Hsiung BK, Lo KM, Doose JM, Thrash JC, Stalder AK, Staufenbiel M, Neumann H, Carson MJ. Dual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice: implications for vaccine-based therapies for Alzheimer's disease. ASN Neuro. 2010 Jul 12;2(3):e00037. PubMed.
Other Citations
External Citations
Further Reading
No Available Further Reading
Primary Papers
- Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Suver C, Shah H, Mahajan M, Gillis T, Mysore J, MacDonald ME, Lamb JR, Bennett DA, Molony C, Stone DJ, Gudnason V, Myers AJ, Schadt EE, Neumann H, Zhu J, Emilsson V. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013 Apr 25;153(3):707-20. PubMed.
Panelists
-
 Eric Schadt
Mount Sinai Hospital
Eric Schadt
Mount Sinai Hospital
-
 Ana Griciuc, Ph.D.
Massachusetts General Hospital and Harvard Medical School
Ana Griciuc, Ph.D.
Massachusetts General Hospital and Harvard Medical School
-
 Monica Carson
University of California, Riverside
Monica Carson
University of California, Riverside
-
 John Hardy, Ph.D.
Institute of Neurology, UCL
John Hardy, Ph.D.
Institute of Neurology, UCL
Current Scientific Advisory Board Member -
 Jeremy Miller, Ph.D.
Allen Institute for Brain Science
Jeremy Miller, Ph.D.
Allen Institute for Brain Science
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.