Transcription Factor Nourishes Neuron-Muscle Pair in ALS Model
Quick Links
A gene that was once considered a simple cell stress marker turns out to be a short-term savior for motor neurons in a mouse model of amyotrophic lateral sclerosis. In the January 13 Proceedings of the National Academy of Sciences online, researchers from Children’s Hospital in Boston report that activating transcription factor 3 (ATF3), which remains dormant in healthy neurons, extends neuron survival and promotes re-innervation of neuromuscular junctions. In mice, this protected against muscle atrophy, though it only brought temporary improvement in muscle strength and a slight delay in disease onset.
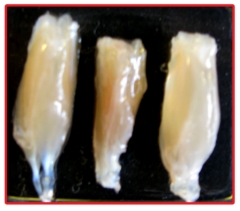
Preventing muscle atrophy. Image courtesy of Rhona Seijffers.
Because SOD1 mice express ATF3 weakly and only in a subset of motor neurons, Seijffers crossed them with a line that constitutively expresses the protein in all mature neurons. Overall, ATF3 delayed but did not prevent pathology. While SOD1 mutants lost 54 and 81 percent of their motor neurons by 90 days and 120 days of age, respectively, the crosses lost only 21 percent and 42 percent at the same ages. The transcription factor also saved axons. While SOD1 mice showed axonal damage as early as 60 days, before symptoms appeared, Seijffers observed no axon damage in the crosses until 120 days.
Because axons will only do a body good if they innervate a muscle, Seijffers examined neuromuscular junctions in the lower hind legs of the crosses. She stained sections for axons and junctions and counted how many were connected. In the standard ALS model, denervation of the gastrocnemius muscle had already begun at 60 days, with 30 percent of junctions lost. By 120 days, that number climbed to 43 percent. In contrast, the crosses maintained full innervation until at least 90 days; at 120 days, 83 percent of the junctions remained innervated. The muscles of mSOD1 mice also wasted away. Compared to wild-type littermates, mSOD1 mice lost 24 percent of their calf-muscle mass by 60 days, and 56 percent by 120 days. The mSOD1/ATF3 mice maintained muscle mass until 90 days, and by 120 days had lost only 20 percent of their calf muscle (see image above).
The researchers noticed an apparent disconnect between the motor neuron survival data and the innervation numbers—one-fifth of the motor neurons in the mSOD1/ATF3 mice died by 90 days, but muscles remained fully innervated at that time. Seijffers interpreted this as a hint that as some neurons die, the remaining neurons sprout new axons and establish new connections to the muscle junctions. When she examined neurons at 120 days, she observed the mSOD/ATF3 crosses sprouted new axons nearly four times as often as the mSOD1 mice.
How did all this activity affect the mice’s ability to move and survive? Not well. Even though the mSOD/AFT3 mice maintained muscle mass and innervation for months longer than the mSOD mice, the crosses retained hind-limb muscle grip strength for only 11 days longer. The study authors suggested that the new axons innervating the muscles were able to prevent atrophy, but unable to stimulate the muscle to contract at full strength. In addition, once the mSOD1/ATF mice developed disease they went downhill at a rate similar to their mSOD1 cousins, surviving for an average of only eight days longer. This extension was “very modest,” commented Flint Beal of Weill Medical College of Cornell University in New York, who noted that better treatments extend life in the same mouse model by nearly three weeks.
How does ATF3 preserve the neurons? In a microarray analysis of gene expression, Seijffers found that the transcription factor activates many pro-survival and regeneration genes. It downregulated pathways involved in apoptosis, and promoted expression of genes involved in neurogenesis, axon myelination, and impulse transmission. Seijffers sees ATF3 as a “master regulator” of neuroprotection. If physicians could boost ATF3 activity in the neurons of people with ALS, perhaps via gene therapy or a drug, it could have broadly beneficial effects, she speculated.
Jeffrey Jasper of Cytokinetics, Inc., of South San Francisco, California, praised the paper but was skeptical of a broadly acting ATF3-based therapy. “I would be scared to do something that would activate so many genes,” he said. “I think if they can tease out which protein or proteins are the most important to get these beneficial effects, that would be a better approach.”
Seijffer’s work indicates that while ATF3 was helpful, it was ultimately insufficient. Her findings fit with another study of muscle-protective therapy in ALS mice, which kept the mice active but did not improve survival (see May 2012 news story). Together, these experiments indicate that neurons and their axons, not muscle, are the primary site of ALS damage, Beal said. Any successful ALS treatment will have to help both the neuron cell body and the neurites reaching toward muscles, Seijffers added.—Amber Dance
References
News Citations
Paper Citations
- Seijffers R, Allchorne AJ, Woolf CJ. The transcription factor ATF-3 promotes neurite outgrowth. Mol Cell Neurosci. 2006 May-Jun;32(1-2):143-54. Epub 2006 May 19 PubMed.
- Seijffers R, Mills CD, Woolf CJ. ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J Neurosci. 2007 Jul 25;27(30):7911-20. PubMed.
Further Reading
Papers
- Vlug AS, Teuling E, Haasdijk ED, French P, Hoogenraad CC, Jaarsma D. ATF3 expression precedes death of spinal motoneurons in amyotrophic lateral sclerosis-SOD1 transgenic mice and correlates with c-Jun phosphorylation, CHOP expression, somato-dendritic ubiquitination and Golgi fragmentation. Eur J Neurosci. 2005 Oct;22(8):1881-94. PubMed.
- Vinciguerra M, Musaro A, Rosenthal N. Regulation of muscle atrophy in aging and disease. Adv Exp Med Biol. 2010;694:211-33. PubMed.
- Steinacker P, Hawlik A, Lehnert S, Jahn O, Meier S, Görz E, Braunstein KE, Krzovska M, Schwalenstöcker B, Jesse S, Pröpper C, Böckers T, Ludolph A, Otto M. Neuroprotective function of cellular prion protein in a mouse model of amyotrophic lateral sclerosis. Am J Pathol. 2010 Mar;176(3):1409-20. PubMed.
- Kliem MA, Heeke BL, Franz CK, Radovitskiy I, Raore B, Barrow E, Snyder BR, Federici T, Kaye Spratt S, Boulis NM. Intramuscular administration of a VEGF zinc finger transcription factor activator (VEGF-ZFP-TF) improves functional outcomes in SOD1 rats. Amyotroph Lateral Scler. 2011 Sep;12(5):331-9. PubMed.
Primary Papers
- Seijffers R, Zhang J, Matthews JC, Chen A, Tamrazian E, Babaniyi O, Selig M, Hynynen M, Woolf CJ, Brown RH Jr. ATF3 expression improves motor function in the ALS mouse model by promoting motor neuron survival and retaining muscle innervation. Proc Natl Acad Sci U S A. 2014 Jan 28;111(4):1622-7. Epub 2014 Jan 13 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Cytokinetics
This is an interesting manuscript. The authors demonstrated that transgenic overexpression of ATF3 in motor neurons of SOD1 mice promoted motor neuron survival and maintained axonal integrity and neuromuscular junction (NMJ) innervations. They found in ATF3/SOD1 mice compared to SOD1 transgenic that a) motor neuron survival was higher, b) there was a reduction in the percentage of large-caliber axons and an increase in the percentage of small-caliber axons, c) the percentage of NMJ innervations was higher, d) muscle atrophy was decreased, and e) ATF3 overexpression slowed onset of disease and slightly prolonged survival (by 7.7 days). ATF3 expression alone was not sufficient to halt disease progression. Importantly, ATF3 overexpression only slowed loss of grip strength by about 10 days. Thus, the nascent collaterally innervated NMJs appear to inhibit muscle atrophy but do not sufficiently maintain muscle strength. Increasing ATF3 expression is an interesting approach to promote motor neuron survival and axonal growth but will likely need to be coupled with other therapeutics to slow/halt the progression of ALS and improve muscle function in patients.
Make a Comment
To make a comment you must login or register.