Seeing Is Believing—Plaque Growth Is Slow, Tapers With Age
Quick Links
To understand how amyloid plaques, the troublesome clumps of Aβ, form and grow in the brain, it helps to just watch the action directly. Advances in multiphoton microscopy have made this possible, allowing scientists to see individual plaques develop within the brains of living mice in real time. Researchers in Germany have now refined the technique to track plaque formation and growth in AD mouse models for up to six months—the longest ever recorded. Two new studies show that plaques grow slowly over time and reach a plateau in older, amyloid-laden animals. The new insight into plaque formation dynamics could enable better monitoring of amyloid-lowering compounds in preclinical development.
The first study, led by Mathias Jucker at the University of Tübingen, Germany, appears in this week’s Journal of Neuroscience. The second study, by Jochen Herms, Ludwig-Maximilians University, Munich, and colleagues was posted December 6 online in Acta Neuropathologica.
A few specialized labs have used in vivo multiphoton microscopy to peer into the brains of AD transgenic mice and track plaque growth through tiny cranial windows. Because it is done in live animals, the analysis is painstaking. Scientists typically rely on fluorescent cells and blood vessels as landmarks to realign the field of view for repeated imaging across multiple time points. However, “because the picture is three-dimensional, if your angle is slightly off, your perspective on the plaque changes,” said Jin-Moo Lee of Washington University School of Medicine in St. Louis, Missouri, who has used two-photon imaging to study plaque dynamics in AD mice, but was not involved in the recent studies.
In Jucker’s paper, first authors Jasmin Hefendehl and Bettina Wegenast-Braun, and colleagues describe a new head restraint system that allows more efficient and accurate multiphoton analysis. The researchers drill a 4-mm-wide hole into the mouse’s skull, seal it with a coverslip, and cement onto that a custom-built titanium ring that fits precisely into a small notch on a motorized stage. Instead of using cells and vessels as signposts to find previously imaged brain areas, “you can basically press a button that does that automatically,” Hefendehl told ARF.
Between imaging sessions, the mice scamper freely, and their behavior and lifespan appear unaffected by the titanium structure fixed to their heads. “We have some mice that have lived quite happily with the ring for two years,” Hefendehl said. Mice can be fitted with the rings once their bone structure is calcified—as early as three months of age—she noted.
Using the new method, which will be described in an upcoming issue of Nature Protocols, the researchers watched plaques develop in the brains of APP/PS1 mice starting at three to four months of age, when amyloid pathology is rampant in this strain. Jucker and colleagues imaged over a 25-week period—a record thus far for multiphoton microscopy studies of AD mice. Imaging weekly for the first two months, biweekly for the next month, and monthly up to 25 weeks, the scientists studied nearly 400 plaques in seven mice, each mouse analyzed at six locations. They observed a period where new plaques form rapidly and grow steadily, and a later phase where the number of new plaques forming peters off.
To analyze the dynamics of this process, the scientists first measured the size distribution of plaques in mice of increasing age (four, seven, and nine months). Four-month-old animals had mostly small plaques of less than 16 micrometers across, whereas plaque size shifted toward larger (up to 44 micrometers diameter) in older mice. Consistent with their predominance in younger animals with less advanced disease, new plaques were small—only 13 percent had a diameter over 8 micrometers, whereas more than three quarters of pre-existing plaques were larger than that.
The team also analyzed changes in volume and diameter for new and already formed plaques. In the volume analysis, new plaques showed higher-fold increases over time, whereas existing plaques showed very little change when assessed in this way. However, when growth was measured in terms of diameter, both types of plaque grew at similar rates. This makes mathematical sense, since a linear increase in diameter would mean that small plaques grow more robustly than large ones when comparing fold increases in volume. “The underlying physiological model suggests that new amyloid ‘layers’ are constantly added to the surface of existing plaques—despite the difference in initial size,” the authors write.
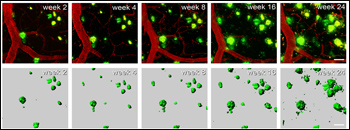
Bulking Up
Long-term multiphoton imaging shows growth of amyloid plaques (green) in the brains of APP/PS1 transgenic mice starting at 3.7 months of age. Blood vessels are stained red. View larger image. Image credit: Hefendehl et al. The Journal of Neuroscience 2011
In the Acta Neuropathologica paper, first author Steffen Burgold and colleagues used a similar approach to visualize plaques in AD mice. The researchers made a small incision in the skull, glued on a coverslip, then attached a small metal bar containing a hole for a screw, to enable repositioning of the mouse for repeated imaging sessions. Compared to Jucker’s team, they analyzed plaque formation over a shorter period (six weeks), and used an AD strain (Tg2576) with slower amyloidosis. Tg2576 mice start accumulating brain amyloid around nine to 10 months.
In weekly imaging sessions, the researchers studied 83 plaques in 12-month-old Tg2576 mice, and found that newborn plaques were, on average, considerably smaller in volume than pre-existing ones, suggesting that plaques take on the order of weeks to mature. Both types grew steadily over time, though relative volumes increased more quickly for newborn than for pre-existing plaques. These patterns corroborate Jucker’s findings in APP/PS1 mice with amyloid pathology well underway. However, older (18-month-old) Tg2576 mice did not form new plaques, and their pre-existing ones hardly increased in volume, over the six-week imaging period.
Overall, the two studies found that new plaques start off small and steadily bulk up before reaching a maximum girth in older mice with more advanced disease. The observation that plaques stop growing in old mice is not new (see Christie et al., 2001). Why that happens remains speculative. In aging mice with “lots of plaques around, soluble Aβ peptides have many choices as to where they can go, making it harder for the growth of any single plaque to be detected,” Herms suggested. As for the age-related decline in the rate of new plaque formation, Hefendehl proposed that it may be a passive process where “existing plaques take up amyloid-β that is floating around. “If there are no plaques to take up the soluble Aβ, then there could be a critical concentration of Aβ present to seed a new plaque,” she said.
The current data contrast with the results of a multiphoton microscopy study by Brad Hyman and colleagues at Massachusetts General Hospital in Boston. Imaging plaques in three AD strains, including the Tg2576 mice, the scientists found a significant number of plaques that sprang up overnight with no further growth in the subsequent two-week imaging (Meyer-Luehmann et al., 2008 and ARF related news story). “Some plaques were very big. To think they just appeared in 24 hours was quite amazing,” said Herms said of that study.
Lee and colleagues did their own multiphoton analysis that offered a reason for the apparent lack of plaque growth in the Hyman study. The researchers imaged β amyloid in APP/PS1 transgenic mice and found plaques growing steadily over a period of weeks, not days (Yan et al., 2009 and ARF related news story). However, plaque growth was observed only when the scientists used an alternative method that involved paring the skull to near transparency and imaging through those ultra-thin bone windows. When they drilled all the way through to create open-skull windows, like those used in the Jucker and Herms studies, the WashU team saw considerable gliosis, which they believe stunted further growth of plaques that formed at early timepoints. A recent Nature Methods paper describes a new approach that improves the durability of thin-skull windows, and discusses the ongoing controversy around procedure-induced gliosis (Drew et al., 2010).
In their recent study, Herms and colleagues averted potential inflammatory responses associated with the open-skull procedure by waiting three weeks after surgery to begin imaging. Jucker’s group delayed imaging for just one week, but detected no gliosis—or in this case, proliferation of microglia expressing green fluorescent protein. Staining mouse brains for activated microglia (Iba-1) and astrocytes (GFAP) a week after the open-skull procedure also found no differences between surgically treated mice and control animals. In addition, the Jucker lab optimized the surgery so the skull can be lifted carefully without disrupting the dura matter. “By now, up to 90 percent of the craniotomies we do produce no gliosis,” noted Wegenast-Braun.
Regardless of procedural differences, scientists cite imaging duration as the prime reason for the apparent discrepancy between the plaque growth dynamics seen in Hyman’s study versus other reports. In an e-mail to ARF, Jucker noted that he thinks his new data “do not nullify” Hyman’s findings. “It just extends his work by showing that amyloid plaques grow over time if imaging is done over a long period,” Jucker wrote. Melanie Meyer-Luehmann, first author on the Hyman paper, agrees, noting that the two present studies “show very nicely that plaques can indeed appear very quickly and therefore confirm what we previously published in three different APP transgenic lines.” Before joining the Hyman lab as a postdoc, Meyer-Luehmann did her Ph.D. studies under Jucker, and now heads her own research group at Ludwig-Maximilians University in Munich.
Taken together, the current work strengthens an emerging consensus in multiphoton imaging studies—plaques can initially appear quickly, then grow slowly over long periods and taper in aging animals with extensive amyloid pathology. “There are now three independent groups that have confirmed this using different techniques and different animal models,” Lee noted. By helping to flesh out the growth dynamics of new and existing Aβ plaques, the data may improve future preclinical testing of potential amyloid-lowering agents for AD and other diseases.—Esther Landhuis
References
News Citations
- Popcorn Plaque? Alzheimer Disease Is Slow, Yet Plaque Growth Is Fast
- Plaque Growth Gradual, Slowed by Gliosis and Aβ Removal
Paper Citations
- Christie RH, Bacskai BJ, Zipfel WR, Williams RM, Kajdasz ST, Webb WW, Hyman BT. Growth arrest of individual senile plaques in a model of Alzheimer's disease observed by in vivo multiphoton microscopy. J Neurosci. 2001 Feb 1;21(3):858-64. PubMed.
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008 Feb 7;451(7179):720-4. PubMed.
- Yan P, Bero AW, Cirrito JR, Xiao Q, Hu X, Wang Y, Gonzales E, Holtzman DM, Lee JM. Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J Neurosci. 2009 Aug 26;29(34):10706-14. PubMed.
- Drew PJ, Shih AY, Driscoll JD, Knutsen PM, Blinder P, Davalos D, Akassoglou K, Tsai PS, Kleinfeld D. Chronic optical access through a polished and reinforced thinned skull. Nat Methods. 2010 Dec;7(12):981-4. PubMed.
Other Citations
Further Reading
Papers
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008 Apr 16;28(16):4283-92. PubMed.
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008 Feb 7;451(7179):720-4. PubMed.
Primary Papers
- Hefendehl JK, Wegenast-Braun BM, Liebig C, Eicke D, Milford D, Calhoun ME, Kohsaka S, Eichner M, Jucker M. Long-term in vivo imaging of β-amyloid plaque appearance and growth in a mouse model of cerebral β-amyloidosis. J Neurosci. 2011 Jan 12;31(2):624-9. PubMed.
- Burgold S, Bittner T, Dorostkar MM, Kieser D, Fuhrmann M, Mitteregger G, Kretzschmar H, Schmidt B, Herms J. In vivo multiphoton imaging reveals gradual growth of newborn amyloid plaques over weeks. Acta Neuropathol. 2011 Mar;121(3):327-35. PubMed.
- Yan P, Bero AW, Cirrito JR, Xiao Q, Hu X, Wang Y, Gonzales E, Holtzman DM, Lee JM. Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J Neurosci. 2009 Aug 26;29(34):10706-14. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Lund University
This is an excellent study by Lee and colleagues employing serial multiphoton microscopy (MPM) to provide more clues to the process of plaque formation in the living brain of a well-established APP/PS1 transgenic mouse model of Alzheimer disease. Previous work by Hyman and colleagues had provided novel observations on the remarkably rapid appearance of plaques, and had also noted that once formed, there was little additional growth in the size of plaques. The focus of the current study is less the appearance and more the growth in the size of existing plaques over a time frame of a few weeks using a thinned skull window approach. They provide intriguing evidence for the importance of the type of window used to visualize plaques. Specifically, Lee and colleagues show that the open craniotomy with coverslip approach used in previous MPM studies in AD prevents the further growth of plaques and even augments regression of some plaques when compared with the thin-skull method. With the open- but not thin-skull method, there is marked cortical activation of inflammatory cells below the cranial window. These data also provide further evidence for the importance of inflammatory cells in modulating plaque pathology. One wonders whether the different methods have a differential effect on neuritic dystrophy. Interestingly, they also show that in younger but not older mice, γ-secretase inhibition retards formation and growth of new plaques while not effecting existing plaques. They suggest that these data support the importance of early rather than later therapeutic intervention in AD, although one can note that AD is also an anatomically progressive disease; less vulnerable brain regions may be at an earlier pathological stage (and therefore more amenable to treatment) than more vulnerable/pathologically advanced brain regions. Additionally, they show that the growth of plaques correlates with extracellular β amyloid levels in the interstitial fluid, a pool of β amyloid that many but not all view as the origin of plaques. Overall, this new MPM study is another important contribution in elucidating the development of β amyloid plaque pathology.
View all comments by Gunnar GourasUniversity of Antwerp
Yan and colleagues add another piece to the plaque kinetics puzzle by showing, with on multiphoton in vivo microscopy, that amyloid plaques in a bigenic PSAPP mouse model appear and grow over a period of weeks before reaching a mature size. These data seem to be in apparent conflict with earlier work using the same technique on related mouse models (Meyer-Luehmann et al. 2008), where dense plaques were shown to reach their maximum size in about a day and thereafter maintain a status quo.
The present study also goes forward to propose a reason for this discrepancy. Amyloid imaging through large open-skull cranial windows (as utilized solely by Meyer-Luehmann and colleagues) seems to activate gliosis, in contrast to thinned-skull windows of ≈1/10th the size, where calvaria are merely thinned down to allow in vivo microscopy without exposing the dura mater. This seems logical, as activation of gliosis has been shown in several studies to be an important factor in limiting plaque growth (Meyer-Luehmann et al. 2008; Bolmont et al., 2008; Yan et al., 2009). The stage of disease also seems to be important, as six-month-old mice with a higher proportion of smaller plaques demonstrate more accelerated plaque growth compared to 12-month-old animals (Yan et al., 2009).
Secondly, however carefully studies attempt to show that the sizes of the plaques estimated by in vivo imaging are true representatives of the plaques occurring at that or a later stage of disease, it is always difficult to do so. Lastly, it’s important to keep in mind that the methoxy-X04 used in multiphoton in vivo microscopy only binds to fibrillar Aβ and not to the soluble/oligomeric forms of Aβ that most likely provide the initial nidus of plaque formation. For this reason I don’t believe that we have had the final word on the kinetics and dynamics of plaque formation. Important from a therapy point of view is that anti-Aβ treatments have to be started as early as possible in order to be efficacious—that everyone agrees on.
References:
Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008 Feb 7;451(7179):720-4. PubMed.
Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008 Apr 16;28(16):4283-92. PubMed.
View all comments by Samir Kumar-SinghUK Dementia Research Institute@UCL and VIB@KuLeuven
This is excellent work. The authors make elegantly the case that the procedures used to visualize amyloid plaques in vivo may strongly affect the generation and dynamics of the plaques. It is also of strong interest that interstitial Aβ peptide is such an important contributor to the plaque dynamics, as this is a rather small pool of total Aβ in the brain, and also highly dynamic and influenced by medication. Finally, the fact that 20-30 percent changes in that pool strongly affect the plaque formation should indeed raise hope that a therapeutic window exists for secretase inhibitors.
I strongly recommend the paper.
View all comments by Bart De Strooper�
In my opinion, the discussion above misses one important fact: Brad Hyman's group published already in 2001 that plaques do not grow over time and that there is a restriction on plaque growth (Christie et al., 2001). In that study, more than 300 plaques were analyzed with two-photon microscopy over a time period of up to five months, and the investigators found the majority of plaques remained unchanged in size over time. Even more importantly, the data were observed using the thinned-skull method, i.e., the same method used by Yan et al., 2009. Therefore, thinned-skull versus open-skull preparation alone cannot account for the opposing result.
References:
Christie RH, Bacskai BJ, Zipfel WR, Williams RM, Kajdasz ST, Webb WW, Hyman BT. Growth arrest of individual senile plaques in a model of Alzheimer's disease observed by in vivo multiphoton microscopy. J Neurosci. 2001 Feb 1;21(3):858-64. PubMed.
View all comments by Melanie Meyer-LuehmannWashington University at St. Louis
We appreciate the comments of Dr. Meyer-Luehmann. However, the absence of plaque growth reported in the Christie et al. (2001) paper is very consistent with the data reported in our recent paper (Yan et al, 2009). Although we observed marked plaque growth in six-month-old APP/PS1 mice (early in plaque pathogenesis), we saw little to no growth in 10-month-old APP/PS1 mice. Of note, the Christie et al. paper did not see plaque growth in 18-month-old (mean age) Tg2576 mice. Therefore, our observations in older animals who have more advanced pathology are in agreement with the Christie et al. paper.
References:
Christie RH, Bacskai BJ, Zipfel WR, Williams RM, Kajdasz ST, Webb WW, Hyman BT. Growth arrest of individual senile plaques in a model of Alzheimer's disease observed by in vivo multiphoton microscopy. J Neurosci. 2001 Feb 1;21(3):858-64. PubMed.
View all comments by Jin-Moo Lee�
This and other research demonstrates the deposition of Aβ in vivo in an animal model. Do we know that the “structures” that are shown being formed are also the same structures that are identified histo- or immunochemically postmortem?
Thus, are we confident that what is observed is the full process that results in the structures that we identify classically as plaques postmortem?
The alternative is that we are observing one part of a process. In some instances what is deposited may eventually be removed or transformed to something else and it is this “something else” which we identify postmortem as senile plaques.
Are senile (neuritic) plaques simply deposits of Aβ, or are they more than this?
View all comments by Chris ExleyAs a graduate student who reviewed this subject in great detail for a journal club (see Meyer-Luehmann et al., 2008 and Yan et al., 2009), I am surprised at some of the opinions presented here after these most recent papers on plaque dynamics (Hefendehl et al., 2011; Burgold et al., 2010), which I think are interesting and thorough examinations of plaque growth in vivo. In contrast, when reviewing the initial paper on this topic from the Hyman Lab (Meyer-Luehmann et al., 2008), it became apparent to me and the people with whom I discussed it that the reason why they saw very rapid plaque appearance and no further plaque growth within 14 days was because of an artifact of incomplete dye labeling. If one inspects in detail Figure 1 in their paper, one can see that the plaque that “appeared” after 24 hours of dye injection was really present even before (just poorly labeled). Consistent with this, the adjacent large plaque seen in the same image underwent a very marked increase in dye labeling within this same interval. This is almost certain to be explained by ongoing dye labeling.
Interestingly, in this same figure, they present their data of all new plaques observed, and coincidentally they all appeared within one day of the first dye injection. This again is consistent with an artifact in which dye labeling is incomplete after 24 hours of initial dye injection. The appearance of a plaque at around 24 hours just reflects the ongoing dye labeling. Incomplete labeling also explains why they did not see any new plaques appearing at any time other than after the first day of dye injection. In my opinion, their paper remains at odds with these more recent papers in the Journal of Neuroscience and Acta Neuropathologica, which show no rapid plaque appearance and report continuous plaque growth over much longer intervals. The lack of growth seen in the Hyman paper is likely to be related to neuroinflammation induced by their imaging procedure as previously demonstrated (Yan et al., 2009).
References:
Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008 Feb 7;451(7179):720-4. PubMed.
Hefendehl JK, Wegenast-Braun BM, Liebig C, Eicke D, Milford D, Calhoun ME, Kohsaka S, Eichner M, Jucker M. Long-term in vivo imaging of β-amyloid plaque appearance and growth in a mouse model of cerebral β-amyloidosis. J Neurosci. 2011 Jan 12;31(2):624-9. PubMed.
Burgold S, Bittner T, Dorostkar MM, Kieser D, Fuhrmann M, Mitteregger G, Kretzschmar H, Schmidt B, Herms J. In vivo multiphoton imaging reveals gradual growth of newborn amyloid plaques over weeks. Acta Neuropathol. 2011 Mar;121(3):327-35. PubMed.
Christie RH, Bacskai BJ, Zipfel WR, Williams RM, Kajdasz ST, Webb WW, Hyman BT. Growth arrest of individual senile plaques in a model of Alzheimer's disease observed by in vivo multiphoton microscopy. J Neurosci. 2001 Feb 1;21(3):858-64. PubMed.
Yan P, Bero AW, Cirrito JR, Xiao Q, Hu X, Wang Y, Gonzales E, Holtzman DM, Lee JM. Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J Neurosci. 2009 Aug 26;29(34):10706-14. PubMed.
Several papers now have used multiphoton imaging to monitor plaques over time in AD transgenic models (Hefendehl et al., 2011; Burgold et al., 2010; Yan et al., 2009), following on the initial work we published in 2001 (Christie et al., 2001). Over the years we have imaged thousands of plaques using either “thin skull” or “coverslip” approaches in three different APP or APP/PS1 overexpressing models. The new papers, emerging from analogous work at Washington University and in Germany, show similar approaches to dissect the natural history of plaques in living animals.
Overall, there is general concurrence in our observations. It is obvious that animals initially have no plaques, then many months later have many plaques. What happens in between? We found that plaques form surprisingly quickly, then reach a near maximal size within days. The other groups, using slightly different models and methods, found that plaques form and then may well continue to grow initially for some time, then reach a plateau where growth ceases. That growth ultimately ceases is obvious—otherwise there would be one large plaque in the brains of elderly mice, and, of course, that is not the case. In fact, postmortem analysis of plaque size distribution reveals no change in the average size of plaques or in the distribution of sizes regardless of age.
Why are there any differences in the observations regarding the slope of the growth of plaques in animal models? Any number of technical issues—ranging from mouse variability to differences in imaging techniques—might help explain the discrepancies. We have measured cross-sectional areas because of the increased resolution of images in the X-Y plane, while other groups use a full Z stack and estimate volume, essentially trading the increased information in the Z stack for the increased uncertainties of the measurements at the top and bottom (given relatively poor Z resolution compared to X-Y resolution in multiphoton optics). Different surgical procedures, different ways of administering dyes, different software packages, or even different optics might impact the subtle analysis of these high-resolution images.
However, the important point is whether any of these observations accurately model what happens in Alzheimer’s disease itself. From this point of view, we have recently completed an analysis of the temporal neocortex of 92 individuals with Alzheimer's disease, and 16 controls, ranging in duration of dementia from six months to almost 20 years. Of course, this is a postmortem histological analysis, so that longitudinal imaging of individual plaques is not possible. Nonetheless, if plaques dramatically grew with increasing duration of illness, we would expect to see evidence of that in the size distribution of either the thioflavin S core or the anti-Aβ immunostained deposits. We found only the most subtle changes over time, with an increase in plaque size over 20 years of ~2 percent per year. We conclude that dramatic continued plaque growth is unlikely to be a central feature of Alzheimer's disease progression, although the conundrum still remains as to why plaques form in the first place, grow to their rather large size, and then presumably ultimately reach a plateau where further growth is inhibited. It may be that careful analysis of what impacts the rate of growth, or of the phenomena that occur after plaques stabilize, will help provide insight. We hope that in vivo multiphoton longitudinal imaging of animal models will continue to help point towards answers to these sorts of questions.
References:
Hefendehl JK, Wegenast-Braun BM, Liebig C, Eicke D, Milford D, Calhoun ME, Kohsaka S, Eichner M, Jucker M. Long-term in vivo imaging of β-amyloid plaque appearance and growth in a mouse model of cerebral β-amyloidosis. J Neurosci. 2011 Jan 12;31(2):624-9. PubMed.
Burgold S, Bittner T, Dorostkar MM, Kieser D, Fuhrmann M, Mitteregger G, Kretzschmar H, Schmidt B, Herms J. In vivo multiphoton imaging reveals gradual growth of newborn amyloid plaques over weeks. Acta Neuropathol. 2011 Mar;121(3):327-35. PubMed.
Yan P, Bero AW, Cirrito JR, Xiao Q, Hu X, Wang Y, Gonzales E, Holtzman DM, Lee JM. Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J Neurosci. 2009 Aug 26;29(34):10706-14. PubMed.
Christie RH, Bacskai BJ, Zipfel WR, Williams RM, Kajdasz ST, Webb WW, Hyman BT. Growth arrest of individual senile plaques in a model of Alzheimer's disease observed by in vivo multiphoton microscopy. J Neurosci. 2001 Feb 1;21(3):858-64. PubMed.
Make a Comment
To make a comment you must login or register.