Research Brief: There’s a Fly in My TDP-43 Research
Quick Links
A fruit fly is joining the menagerie of TDP-43 animal models, bringing the power of Drosophila genetics to the lab bench. Researchers from the Northwestern University School of Medicine in Chicago, Illinois, present the model in a paper posted online by PNAS this week. The flies express wild-type human TDP-43, a protein linked to both amyotrophic lateral sclerosis and frontotemporal dementia. When the human protein is expressed in motor neurons, the animals show axonal swelling, reduced axonal branching, and ultimately motor neuron loss and mobility problems.
This fly joins a mouse (see ARF related news story on Wegorzewska et al., 2009), a rat (see ARF related news story on Tatom et al., 2009), and a host of other TDP-43 animals (see ARF related news story) helping to move TDP-43 research forward.
The current work was led by first author Yan Li and principal investigator Jane Wu. They generated flies expressing TDP-43, hitched to red fluorescent protein (RFP), under neuron-specific promoters. When they expressed the transgene in motor neurons, the flies failed to hatch from the final pupa stage, so they studied the larvae. These moved less than their control counterparts expressing only RFP. Additionally, the researchers created viable adults by turning on the transgene only in adult tissues, allowing the larvae to develop normally. The adult flies also spent less time in motion than did control flies.
TDP-43 localization appears to be key to its pathogenesis, with the normally nuclear protein migrating into the cytoplasm in disease conditions (see ARF related news story on Barmada et al., 2010; Jane Wu is a coauthor on this recent paper). Under the microscope, the researchers saw that most of the TDP-43 transgene remained nuclear, but when it moved into the cytoplasm the cells displayed swelling, nuclear fragmentation, and cell death.
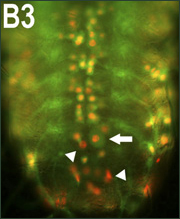
Transgenic Drosophila expressing human TDP-43 (red) in their motor neurons (membranes in green) evince swelling (arrow) and fragmented or condensed nuclei (arrowheads). Image credit: Proceedings of the National Academy of Sciences USA
TDP-43 contains two amino-terminal RNA-binding domains and a carboxyl-terminal glycine-rich region. Both have been linked to toxicity (see ARF related news story on Zhang et al., 2009 and ARF related news story on Seyfried et al., 2010). When Li and colleagues expressed solely the carboxyl-terminal fragment in their flies, they found no neurotoxicity, supporting evidence that the amino terminus contributes to pathogenesis.
Many TDP-43 mutations have been linked to disease; however, TDP-43 proteinopathies also exist in people without mutations. In these flies, surplus wild-type TDP-43 was sufficient to cause problems. If that is so in human disease, then reducing TDP-43 might alleviate the symptoms. The researchers created double mutants carrying the human TDP-43 transgene but lacking Drosophila TDP. These animals moved around more than the animals with excess TDP, suggesting that simply increasing TDP levels is enough to cause pathology.
The authors suggest their model will be useful to identify genes that interact with TDP-43 and to test drugs that might dampen the protein’s toxic effects.—Amber Dance
References
News Citations
- Meet the First Published TDP-43 Mouse
- TDP-43 Roundup: New Models, New Genes
- London, Ontario: TDP-43 Across the Animal Kingdom at ALS Meeting
- TDP-43: Modified and On the Move
- Toxic TDP-43 Truncates Point to Gain-of-Function Role in Disease
Paper Citations
- Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2009 Nov 3;106(44):18809-14. Epub 2009 Oct 15 PubMed.
- Tatom JB, Wang DB, Dayton RD, Skalli O, Hutton ML, Dickson DW, Klein RL. Mimicking aspects of frontotemporal lobar degeneration and Lou Gehrig's disease in rats via TDP-43 overexpression. Mol Ther. 2009 Apr;17(4):607-13. PubMed.
- Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci. 2010 Jan 13;30(2):639-49. PubMed.
- Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, Lin WL, Tong J, Castanedes-Casey M, Ash P, Gass J, Rangachari V, Buratti E, Baralle F, Golde TE, Dickson DW, Petrucelli L. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 2009 May 5;106(18):7607-12. PubMed.
- Seyfried NT, Gozal YM, Dammer EB, Xia Q, Duong DM, Cheng D, Lah JJ, Levey AI, Peng J. Multiplex SILAC analysis of a cellular TDP-43 proteinopathy model reveals protein inclusions associated with SUMOylation and diverse polyubiquitin chains. Mol Cell Proteomics. 2010 Apr;9(4):705-18. PubMed.
Further Reading
Papers
- Del Bo R, Ghezzi S, Corti S, Pandolfo M, Ranieri M, Santoro D, Ghione I, Prelle A, Orsetti V, Mancuso M, Sorarù G, Briani C, Angelini C, Siciliano G, Bresolin N, Comi GP. TARDBP (TDP-43) sequence analysis in patients with familial and sporadic ALS: identification of two novel mutations. Eur J Neurol. 2009 Jun;16(6):727-32. Epub 2009 Feb 19 PubMed.
- Moisse K, Mepham J, Volkening K, Welch I, Hill T, Strong MJ. Cytosolic TDP-43 expression following axotomy is associated with caspase 3 activation in NFL-/- mice: support for a role for TDP-43 in the physiological response to neuronal injury. Brain Res. 2009 Nov 3;1296:176-86. PubMed.
- Corrado L, Ratti A, Gellera C, Buratti E, Castellotti B, Carlomagno Y, Ticozzi N, Mazzini L, Testa L, Taroni F, Baralle FE, Silani V, D'Alfonso S. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum Mutat. 2009 Apr;30(4):688-94. PubMed.
- Geser F, Martinez-Lage M, Robinson J, Uryu K, Neumann M, Brandmeir NJ, Xie SX, Kwong LK, Elman L, McCluskey L, Clark CM, Malunda J, Miller BL, Zimmerman EA, Qian J, Van Deerlin V, Grossman M, Lee VM, Trojanowski JQ. Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol. 2009 Feb;66(2):180-9. PubMed.
- Kim SH, Shi Y, Hanson KA, Williams LM, Sakasai R, Bowler MJ, Tibbetts RS. Potentiation of amyotrophic lateral sclerosis (ALS)-associated TDP-43 aggregation by the proteasome-targeting factor, ubiquilin 1. J Biol Chem. 2009 Mar 20;284(12):8083-92. PubMed.
- Moisse K, Volkening K, Leystra-Lantz C, Welch I, Hill T, Strong MJ. Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: implications for TDP-43 in the physiological response to neuronal injury. Brain Res. 2009 Jan 16;1249:202-11. PubMed.
News
- New Ubiquitinated Inclusion Body Protein Identified
- TDP-43 Roundup: New Models, New Genes
- TDP-43: Modified and On the Move
- Meet the First Published TDP-43 Mouse
- All Shook Up: TDP-43 an Instigator of Aggregation
- Heady Times for Researchers Studying TDP-43
- ALS Research: More TDP-43, and Peripherin No Longer in Periphery?
- Gene Mutations Place TDP-43 on Front Burner of ALS Research
- London, Ontario: TDP-43 Across the Animal Kingdom at ALS Meeting
- Toxic TDP-43 Truncates Point to Gain-of-Function Role in Disease
Primary Papers
- Li Y, Ray P, Rao EJ, Shi C, Guo W, Chen X, Woodruff EA, Fushimi K, Wu JY. A Drosophila model for TDP-43 proteinopathy. Proc Natl Acad Sci U S A. 2010 Feb 16;107(7):3169-74. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.