Proteins Backpedal, Spreading Neurodegeneration
Quick Links
Amyloid-β can kill neurons whose cell bodies lie far away, but researchers are unsure how. A study in the August 28 Cell from the lab of Ulrich Hengst, Columbia University, New York, suggests that the peptide triggers local synthesis of a protein messenger that travels back along axons to unleash death signals in otherwise healthy cell bodies. Hengst and colleagues finger the transcription factor ATF4 as the deadly harbinger. They found more ATF4 in AD brains than in control tissue, and eliminating the protein in mice reduced the neurodegenerative effects of Aβ.
“We establish intra-axonal protein synthesis as a pathway by which neurodegeneration is transmitted over long distances in the brain,” said Hengst. Tao Ma of Wake Forest University School of Medicine, Winston-Salem, North Carolina, said that this study hints at a new neurotoxic mechanism for Aβ. “These results suggest a signaling pathway downstream of Aβ that explains how the peptide kick-starts neurodegeneration,” Ma said.
For decades, protein synthesis in axons was thought to be restricted to development (for a review, see Piper and Holt, 2004). However, mounting evidence reports that well into adulthood, the axons of mature neurons routinely translate proteins, and that disturbing this process may contribute to diseases such as fragile X syndrome and spinal muscular atrophy (see Dubacq et al., 2009; Willis et al., 2011; and Wang et al., 2007). Protein synthesis may help neurons regenerate, as it ramps up in axons after injury (for a review see Rishal and Fainzilber, 2014).
Hengst and colleagues wondered if, like injury, neurodegenerative stimuli such as the presence of Aβ would spur protein translation, and if so, what role this played in neurodegenerative processes.
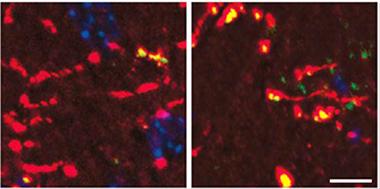
Mouse dentate gyrus axons (red) accumulate more ATF4 protein (green/yellow) when exposed to Aβ42 (right panel), than do control axons (left). Cell bodies in blue. [Image courtesy of Cell, Baleriola et al., Fig. 5.]
To find out, first author Jimena Baleriola and colleagues cultured rat embryonic hippocampal neurons in chambers that separate axons from soma. To axons, they applied synthetic Aβ oligomers to yield a concentration of 250nM in the axonal chamber, in the range reported for AD brain (see Wang et al., 1999). At 24 hours, molecular markers of protein synthesis rose in the axons. After 48 hours, about 40 percent more Aβ-treated neurons exhibited apoptotic markers than did controls, and 13 percent more died as judged by TUNEL staining. If the researchers inhibited protein synthesis or transport back to the cell body, the neurons lived despite the applied Aβ. This hinted that a protein synthesized in the axon traveled back to the soma to cause cell death.
Taking a closer look at the axonal translation, the authors found 151 and 210 mRNAs that were more or less abundant, respectively, when axons were treated with Aβ than in controls. Activating transcription factor 4 (ATF4) stood out as potentially important because it has been previously implicated in suppressing memory-related genes and activating apoptotic ones (see Ameri and Harris, 2008; Ron and Harding, 2012). The researchers found that ATF4 was indeed translated and that the protein traveled back to the cell body. When Baleriola used siRNA in the axon chamber to silence those local ATF4 transcripts, cells stopped dying as a result of Aβ. Inhibiting CHOP, one of the downstream targets of ATF4, also halted neuronal death. Previous studies reported that prolonged expression of CHOP kills cells (see Zinszner et al., 1998).
To see if this mechanism applied in vivo, the researchers next looked to a mouse model of amyloidopathy (see Sotthibundhu et al., 2008). They injected Aβ into the dentate gyrus of wild-type mice, and for a week observed cholinergic neurons that projected to it from the basal forebrain. Over seven days, protein translation revved up in basal forebrain cholinergic axons and ATF4 abounded. The cholinergic neurons expressed CHOP and apoptotic markers, and 20 percent of them died. As in cultured neurons, if the researchers injected the dentate gyrus with siRNA specific for AFT4, basal forebrain cholinergic cells lived.
To look for signs of this process in human disease, the authors analyzed postmortem brains from eight patients with Alzheimer’s disease and eight age-matched controls from the New York Brain Bank at Columbia University. They found ATF4 mRNA and its protein more often in axons of the subiculum and entorhinal cortex of patients compared with controls. The entorhinal cortex is one of the first regions in the brain to accumulate Aβ, and neurons from the subiculum project there.
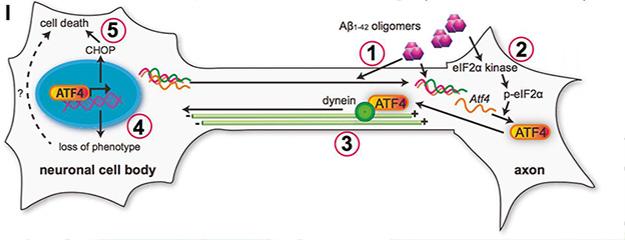
Proposed mechanism of ATF4 involvement in Aβ-induced cell death. Oligomeric Aβ (1) drives local translation of ATF4 (2), which travels (3) back to the soma to drive transcription (4). Activated genes, including CHOP, cause cell death (5). [Image courtesy of Cell, Baleriola et al., Fig. S6.]
According to these new findings, ATF4 and its transport from the axons to the cell body may help explain neurodegeneration in Alzheimer’s that occurs remotely from Aβ, said Hengst (see Liu et al., 2008; Marcyniuk et al., 1986). Researchers should look beyond simple transcription to where mRNAs are translated to fully understand neuropathogenic pathways, he said. ATF4 might be a therapeutic target in AD, he added, pointing out that small molecules can inhibit upstream regulators of ATF4 translation (see Moreno et al., 2013). His group plans to study other mRNAs that turned up in Aβ-treated axons, which include known players in AD such as ApoE, APP, and Clu. The researchers are unsure what their axonal translation means for AD.
The study jibes with a paper that came out last year from the lab of Eric Klann of New York University. First author Tao Ma found that suppressing phosphorylation of eukaryotic initiation factor 2 α-subunit (eIF2α), which reduces translation of ATF4, restored synaptic plasticity and spatial memory in AD transgenic mice (see Ma et al., 2013). “The current study is very consistent with our findings,” said Ma, who now heads his own lab at Wake Forest. He noted that behavioral studies in transgenic mice would be a logical next step to determine if quenching ATF4 translation rescues memory or other behavioral phenotypes.
Gunnar Gouras of Lund University, Sweden, cautioned that this mechanism does not yet point to a solid therapeutic route. ATF4 has a normal function in the neuron, and inhibiting it likely comes with unintended effects, he said. He noted that researchers need to work out how Aβ leads to an uptick in axonal ATF4. Nevertheless, he called Hengst's work elegant, praising the use of techniques that are relatively new for the Alzheimer’s research field. —Gwyneth Dickey Zakaib
References
Paper Citations
- Dubacq C, Jamet S, Trembleau A. Evidence for developmentally regulated local translation of odorant receptor mRNAs in the axons of olfactory sensory neurons. J Neurosci. 2009 Aug 19;29(33):10184-90. PubMed.
- Willis DE, Xu M, Donnelly CJ, Tep C, Kendall M, Erenstheyn M, English AW, Schanen NC, Kirn-Safran CB, Yoon SO, Bassell GJ, Twiss JL. Axonal Localization of transgene mRNA in mature PNS and CNS neurons. J Neurosci. 2011 Oct 12;31(41):14481-7. PubMed.
- Wang W, van Niekerk E, Willis DE, Twiss JL. RNA transport and localized protein synthesis in neurological disorders and neural repair. Dev Neurobiol. 2007 Aug;67(9):1166-82. PubMed.
- Rishal I, Fainzilber M. Axon-soma communication in neuronal injury. Nat Rev Neurosci. 2014 Jan;15(1):32-42. Epub 2013 Dec 11 PubMed.
- Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol. 1999 Aug;158(2):328-37. PubMed.
- Ameri K, Harris AL. Activating transcription factor 4. Int J Biochem Cell Biol. 2008;40(1):14-21. Epub 2007 Jan 28 PubMed.
- Ron D, Harding HP. Protein-folding homeostasis in the endoplasmic reticulum and nutritional regulation. Cold Spring Harb Perspect Biol. 2012 Dec 1;4(12) PubMed.
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998 Apr 1;12(7):982-95. PubMed.
- Sotthibundhu A, Sykes AM, Fox B, Underwood CK, Thangnipon W, Coulson EJ. Beta-amyloid(1-42) induces neuronal death through the p75 neurotrophin receptor. J Neurosci. 2008 Apr 9;28(15):3941-6. PubMed.
- Liu Y, Yoo MJ, Savonenko A, Stirling W, Price DL, Borchelt DR, Mamounas L, Lyons WE, Blue ME, Lee MK. Amyloid pathology is associated with progressive monoaminergic neurodegeneration in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2008 Dec 17;28(51):13805-14. PubMed.
- Marcyniuk B, Mann DM, Yates PO. The topography of cell loss from locus caeruleus in Alzheimer's disease. J Neurol Sci. 1986 Dec;76(2-3):335-45. PubMed.
- Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, Mallucci GR. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med. 2013 Oct 9;5(206):206ra138. PubMed.
- Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, Cavener DR, Klann E. Suppression of eIF2α kinases alleviates Alzheimer's disease-related plasticity and memory deficits. Nat Neurosci. 2013 Sep;16(9):1299-305. PubMed.
External Citations
Further Reading
Primary Papers
- Baleriola J, Walker CA, Jean YY, Crary JF, Troy CM, Nagy PL, Hengst U. Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell. 2014 Aug 28;158(5):1159-72. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Wisconsin
This Cell paper by Baleriola and colleagues describes how oligomeric Aβ1-42 can trigger the rapid recruitment and local translation of a defined set of mRNAs, including that for transcription factor ATF4, and AD-related messages such as APP in axons. Their evidence for the posttranscriptional regulation of locally translated messages in axons dovetails nicely with recent studies in the fragile X field regarding the presynaptic location and function of FMRP (see Akins et al., 2009, and 2012). FMRP is an RNA-binding protein that is found in both pre- and postsynaptic compartments and functions in protein synthesis-dependent long-term plasticity (see Till et al., 2010) as well as activity-dependent axon pruning (Tessier and Broadie, 2008). FMRP represses the translation of numerous synaptic mRNAs, including APP mRNA (see Westmark and Malter, 2007). APP is cleaved by β- and γ-secretases to generate Aβ, which has been proposed to act as a positive regulator of synaptic transmission presynaptically and a negative regulator postsynaptically (Palop and Mucke, 2010). The authors’ novel findings regarding axonal translation of APP suggest that both dendritic and axonal components of the synapse are important in maintaining homeostatic levels of APP and Aβ and contribute to a growing body of evidence supporting an Aβ-mediated feedback mechanism that regulates the synthesis of APP, likely through an FMRP- and mGluR5-dependent pathway.
References:
Akins MR, Berk-Rauch HE, Fallon JR. Presynaptic translation: stepping out of the postsynaptic shadow. Front Neural Circuits. 2009;3:17. Epub 2009 Nov 4 PubMed.
Akins MR, Leblanc HF, Stackpole EE, Chyung E, Fallon JR. Systematic mapping of fragile X granules in the mouse brain reveals a potential role for presynaptic FMRP in sensorimotor functions. J Comp Neurol. 2012 Nov 1;520(16):3687-706. PubMed.
Till SM, Li HL, Miniaci MC, Kandel ER, Choi YB. A presynaptic role for FMRP during protein synthesis-dependent long-term plasticity in Aplysia. Learn Mem. 2011 Jan;18(1):39-48. Print 2011 Jan PubMed.
Tessier CR, Broadie K. Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Development. 2008 Apr;135(8):1547-57. Epub 2008 Mar 5 PubMed.
Westmark CJ, Malter JS. FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 2007 Mar;5(3):e52. PubMed.
Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010 Jul;13(7):812-8. PubMed.
View all comments by Cara WestmarkBarrow Neurological Institute
What is most striking in this work is the observation that Aβ initiates axonal synthesis, a process that has been discussed in other systems. Aβ induces the axonal synthesis of the transcription factor ATF4. The authors use a microfluidic chamber to isolate hippocampal axons from their cell bodies. As with many studies of AD-related molecular pathologic mechanistic findings, the initial studies are performed using embryonic hippocampal neurons, which do not replicate either the aged or diseased microneuronal environment.
Intriguingly, the authors were able to induce retrograde degeneration in select cholinergic neuronal basal forebrain subfields mainly within the nucleus of the diagonal band following Aβ-induced ATF4 transcriptional activity within hippocampal cholinergic projection axons. This degeneration could be blocked by co-injecting Atf4 siRNA into the mice. The selective effect of Aβ42 and ATF4 synthesis on cholinergic neurons of the diagonal band, and not the medial septum, in mice is fascinating. It is possible that not all cholinergic neurons respond to ATF4-induced neurodegenerative signaling, or, as hinted at by the authors, that ATF4 pathobiology may only induce a phenotypic downregulation of neurotransmitter expression in select cholinergic neuronal populations. This is possible but would be a bit surprising since both cholinergic subfields provide the major cholinergic innervation to the hippocampus.
It would have been interesting to know what division of the nucleus of the diagonal band the authors were referring to in their paper. One assumes it was the vertical limb of the diagonal band. The authors also report ATF4-containing axonal structures in the subiculum and the entorhinal cortex in human AD and control cases, with a higher frequency in the AD entorhinal cortex. This is interesting since they did not find ATF4 pathology in the hippocampus, which receives a major glutaminergic innervation pathway from the entorhinal cortex. It is this pathway that is disconnected early in AD. If ATF4 was a factor in retrograde induced degeneration, why did the authors not find ATF4 in the human hippocampus? In this regard, despite their control cases displaying moderate Braak pathology, they did not find ATF4 in these cases either. Perhaps the ATF4 retrograde neurodegenerative signaling pathway does not explain the mechanism underlying neuronal degeneration in human AD or plays a reduced role compared to that seen in mice.
View all comments by Elliott MufsonMake a Comment
To make a comment you must login or register.