No Metal, No Stability: Structure of Apo SOD1
Quick Links
Without its associated metal ions, copper-zinc superoxide dismutase 1 (SOD1) adopts a loose, fluid structure that exposes normally buried cysteines, allowing it to link up with other SOD1 molecules via disulfide bridges. SOD1, which is mutated in one-fifth of people with familial amyotrophic lateral sclerosis, forms aggregates in those people and in animal models of the disease. In a paper published online this week in PNAS, scientists report on the structure of both wild-type and mutant Apo SOD1, and find that the metal-free state is disordered in solution. The data further support the idea that Apo SOD1 is responsible for aggregation of the protein.
Previously, Lucia Banci, Ivano Bertini, and colleagues at the University of Florence, Italy, showed that both wild-type and mutant SOD1 undergo oligomerization only when in the non-metallated state (Banci et al., 2008). “In this paper, the reason for the dramatically different behavior of Apo and metallated forms of SOD1 is now understood,” Bertini wrote in an e-mail to ARF. In mature, metal-bound SOD1, cysteines 6 and 111 are buried deep within the protein. But in metal-free SOD1, the two cysteines rise to the surface where they can interact with cysteines on other Apo SOD1 molecules and seed oligomerization.
The scientists used both NMR and X-ray diffraction to analyze the structures of wild-type SOD1 and two mutants: T54R and I113T. In crystal form, the structures of all three were nearly identical. In solution, the Apo form of SOD1 was disordered, showing a variety of conformations. The loops connecting the protein’s β-strands were particularly flexible. The holo form, in contrast, maintains a stable structure.
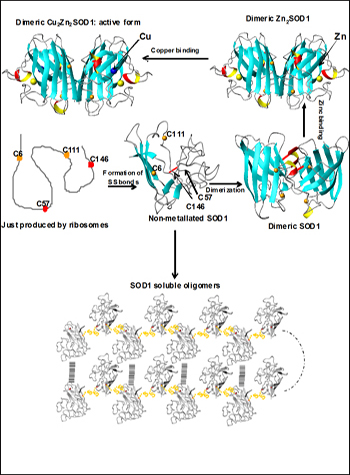
Once translated, SOD1 must form an intramolecular disulfide bridge, dimerize, and pick up both copper and zinc ions in order to mature to the active form (top). Banci et al. propose that the immature, non- metallated form may oligomerize via disulfide bonds between normally buried cysteines (bottom). Image credit: Ivano Bertini, University of Florence (View larger image)
Importantly, the scientists did not use a thermostable SOD1 mutant common in structural studies, noted Jeffrey Agar of Brandeis University in Waltham, Massachusetts, who was not involved with the work. Many researchers find a SOD1 mutant without cysteines 6 and 111 easier to work with, but that mutant doesn’t cause ALS, Agar said, so has limited relevance to the disease. “I hope that other biophysical people take notice” and use the native protein, too, he said.
Banci and colleagues suggest that the metal-free form of SOD1 is key to seeding aggregation. “The mutation is not necessarily the cause of the disease, but more likely the absence of metal is,” Bertini wrote. In that case, the authors surmise, a SOD1 mutation isn’t absolutely necessary to put the protein on a pathological path. “As a consequence, the soluble oligomeric species might represent the precursor toxic species, suggesting a common mechanism for ALS and FALS,” Bertini wrote.
“I think what they’ve done with SOD1 is very pretty. It certainly tells us more about the protein itself,” said Gregory Petsko, also at Brandeis University. However, he said, “I would hesitate to draw as general a set of conclusions as these authors seem to draw.” Mutant SOD1 accounts for only a fraction of FALS cases, and those inherited forms of the disease are vastly outnumbered by sporadic cases. In most people with ALS, TDP-43-containing aggregates, not SOD1 clusters, are the norm. And even among those with SOD1-based ALS, there are more than 100 mutations in the protein that can cause disease. So to extrapolate from a few SOD1 forms to the rest of ALS, Petsko suggested, is a bit of a stretch.—Amber Dance
References
Paper Citations
Other Citations
{kind=link}
Further Reading
Papers
- Chattopadhyay M, Durazo A, Sohn SH, Strong CD, Gralla EB, Whitelegge JP, Valentine JS. Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc Natl Acad Sci U S A. 2008 Dec 2;105(48):18663-8. PubMed.
- Banci L, Bertini I, Durazo A, Girotto S, Gralla EB, Martinelli M, Valentine JS, Vieru M, Whitelegge JP. Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: a possible general mechanism for familial ALS. Proc Natl Acad Sci U S A. 2007 Jul 3;104(27):11263-7. PubMed.
- Di Noto L, Whitson LJ, Cao X, Hart PJ, Levine RL. Proteasomal degradation of mutant superoxide dismutases linked to amyotrophic lateral sclerosis. J Biol Chem. 2005 Dec 2;280(48):39907-13. PubMed.
- Arnesano F, Banci L, Bertini I, Martinelli M, Furukawa Y, O'Halloran TV. The unusually stable quaternary structure of human Cu,Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status. J Biol Chem. 2004 Nov 12;279(46):47998-8003. PubMed.
- Elam JS, Taylor AB, Strange R, Antonyuk S, Doucette PA, Rodriguez JA, Hasnain SS, Hayward LJ, Valentine JS, Yeates TO, Hart PJ. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat Struct Biol. 2003 Jun;10(6):461-7. PubMed.
Primary Papers
- Banci L, Bertini I, Boca M, Calderone V, Cantini F, Girotto S, Vieru M. Structural and dynamic aspects related to oligomerization of apo SOD1 and its mutants. Proc Natl Acad Sci U S A. 2009 Apr 28;106(17):6980-5. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Keio University
This study characterizes the dynamic behavior of SOD1 in detail. First, it essentially reproduces previous studies including the ones from the authors' group, as it has been well known that overall structures are similar between wild-type and mutant SOD1 proteins. In addition, significant differences in the dynamic behavior have been observed between Apo and holo forms of SOD1. When the metal ions are removed from the protein, structural disorder increases particularly in the loop regions.
We think that one of the interesting findings in this paper is the increased solvent accessibility of Cys-6 upon metal removal. Cys-6 is one of the four Cys residues (Cys-6, 57, 111, 146) in SOD1 and is buried toward the protein interior in the holo form of SOD1. In an enzymatically active form of SOD1, an intra-molecular disulfide forms between Cys-57 and 146, while Cys-6 and 111 remain reduced. In contrast, pathological inclusions purified from several ALS-model mice contain SOD1 multimers that are cross-linked via non-physiological disulfide bonds (Furukawa et al., 2006).
It is, however, still controversial which Cys residues are involved in the formation of cross-linked SOD1 multimers under pathological conditions. While we have previously reported that the disulfide formation is not absolutely required for triggering SOD1 aggregation (Furukawa et al., 2008), an important role of Cys-6 and 111 in the formation of disulfide cross-links has been also suggested in the cultured cell model (Niwa et al., 2007). In addition, ALS-causing mutations at position 6 have been reported (i.e., C6G and C6F), implying that the other Cys residues are involved in the formation of disulfide-linked multimers even when Cys-6 is unavailable for disulfide formation. Nonetheless, the increased flexibility and solvent accessibility of Cys-6 upon metal removal will be an important clue to explain a molecular mechanism of the pathological SOD1 oligomer formation.
References:
Furukawa Y, Fu R, Deng HX, Siddique T, O'Halloran TV. Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide dismutase aggregates in spinal cords of model mice. Proc Natl Acad Sci U S A. 2006 May 2;103(18):7148-53. PubMed.
Furukawa Y, Kaneko K, Yamanaka K, O'Halloran TV, Nukina N. Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in the familial form of amyotrophic lateral sclerosis. J Biol Chem. 2008 Aug 29;283(35):24167-76. PubMed.
Niwa J, Yamada S, Ishigaki S, Sone J, Takahashi M, Katsuno M, Tanaka F, Doyu M, Sobue G. Disulfide bond mediates aggregation, toxicity, and ubiquitylation of familial amyotrophic lateral sclerosis-linked mutant SOD1. J Biol Chem. 2007 Sep 21;282(38):28087-95. PubMed.
Make a Comment
To make a comment you must login or register.