Averting a Late-life Crisis: Midlife ApoE2 Clears Plaques in Mice
Quick Links
ApoE’s complex links to Aβ pathology have engendered debate about whether modulating this lipoprotein makes for a rational treatment strategy in Alzheimer’s disease. Animal studies show that ApoE, depending on the isoform, either heightens or lowers AD risk from birth, but scientists are unsure if tweaking ApoE later in life will do the same. A report in the November 20 Science Translational Medicine suggests it may. Scientists led by Bradley Hyman, Massachusetts General Hospital, Charlestown, found that introducing the protective human ApoE2 isoform into the brains of older mice cleared amyloid plaques that were already there. ApoE4 had the opposite effect. “By manipulating ApoE, Alzheimer’s disease pathology, at least in mice, can be modified after it has already begun,” Hyman told Alzforum. “Any therapeutic that manipulates ApoE after disease has started would, in principle, be supported by these data.”
Previous studies by David Holtzman's group at the Washington University of Medicine, St. Louis, have suggested that human ApoE4 expression slows clearance of Aβ from mouse brains, while ApoE2 hastens it (see July 2010 conference story). The ApoE4 mice end up with more amyloid, while those with the ApoE2 allele are protected (see Fagan et al., 2002), mimicking the human condition. However, no studies have clued researchers in to whether a change to ApoE initiated in adult animals will affect pathology.
Seeking an answer, first author Eloise Hudry and colleagues injected an adeno-associated viral vector carrying human ApoE isoforms into the lateral ventricles of seven-month-old APPswe/PS1 mice (see Liu et al., 2005). At that age, these animals have rampant amyloid plaques in the brain. Within two months, the virus permeated the cells lining the lateral ventricles, which released human ApoE into the cerebrospinal and interstitial fluid. The protein reached large swaths of the cortex. The mice still expressed their own ApoE, and total levels of the protein rose by about 10 percent. Over the next three months, the researchers monitored the effects of each ApoE allele on Aβ deposition and neurotoxicity.
Compared with the animals that received human ApoE3, those that got ApoE4 generated higher levels of soluble Aβ40 and Aβ42 in their brains and built up more cortical amyloid deposits. In those treated with ApoE2, Aβ levels fell and fewer plaques accumulated (see image below). According to in-vivo two-photon images taken sequentially from the same animal over two months, amyloid plaques formed more quickly in ApoE4-treated animals than in ApoE2-treated animals.
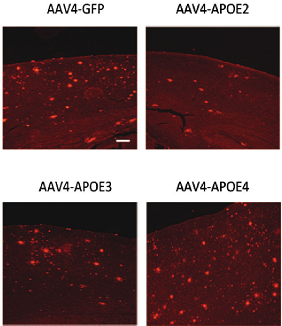
ApoE Modulates Plaques Delivered via the AAV4 vector, ApoE4 isoforms modulate the number of plaques (red) in the cortex of APP/PS1 transgenic mice. Image courtesy of Science Translational Medicine/AAAS.
Looking beyond Aβ, Hudry and colleagues examined neuronal changes surrounding plaques. Array tomography, a high-resolution technique that images immunofluorescence staining on ultrathin tissue slices, revealed that synapses degenerated around plaques in ApoE3- and ApoE4-expressing animals more so than in ApoE2 mice. However, synapses looked no different far away from the deposits, suggesting that any synaptic dysfunction resulted from modulation of Aβ.
In addition, more dystrophic neurites formed near amyloid deposits in ApoE4 animals compared with mice expressing other alleles. To verify their results in a second model, the researchers delivered ApoE to the lateral ventricles of 16- to 18-month-old Tg2576 mice, which already contained plaques. After three months, added ApoE4 led to a higher concentration of soluble Aβ oligomers in the interstitial fluid of the hippocampus than did added ApoE2. Animals receiving ApoE4 also built up more plaques.
Together, the data suggest that ApoE isoforms differentially affect Aβ. How? The researchers looked at Aβ production and clearance, which have long been associated with ApoE. Processing of the amyloid precursor protein occurred to the same extent among the treated mice, suggesting that none of the added ApoE isoforms altered Aβ production. However, the researchers saw lower levels of Aβ40 in the plasma of ApoE4- and ApoE3-expressing mice than controls or mice making ApoE2. That indicates these two alleles slow clearance of Aβ into the blood relative to ApoE2, retaining Aβ in the central nervous system.
The authors have no data yet to suggest whether these Aβ changes translate to learning or memory benefits, but Hudry said they will examine that once they have optimized the system for getting ApoE into the brain.
How do these findings relate to therapy? They imply that ApoE2, or small molecules that mimic it, could serve as candidate AD therapies, wrote the authors. Introducing a large molecule like ApoE2 itself into the brain would be difficult, but small molecules that modulate expression of the lipoprotein are being tested in people. Two clinical trials will examine bexarotene, an approved cancer drug that happens to enhance production of endogenous ApoE, boost its lipidation, and clear plaques in mice (see Feb 2012 news story). Gary Landreth, Case Western Reserve University, Cleveland, has initiated a Phase 1b trial to examine the drug’s effect on Aβ production and clearance in healthy volunteers (see May 2013 news story). Researchers at the Cleveland Clinic in Ohio have begun a Phase 2 trial in patients with mild to moderate AD to see if bexarotene reduces their amyloid burden. Meanwhile, the drug’s impact on plaques in mice has become controversial (see May 2013 news story).
If gene therapy based on injection into the brain ventricles ever proved safe for humans, it could deliver ApoE2 to widespread areas of the cortex, suggested the authors. However, Hyman pointed out that whether ApoE diffuses as well in larger animals as it does in mouse brains remains to be seen. The authors also noted that ApoE2 is known to be more lipidated than ApoE3 and ApoE4, and an increased lipidation state could explain the superior influence on plaque clearance in these AD mouse models. Interestingly, bexarotene is known to boost ApoE lipidation of all ApoE isoforms.
“These experiments are done in adult animals, suggesting that if manipulations like this can be done in the adult human brain, one would want to increase ApoE2 and decrease ApoE4 for therapeutic efficacy,” Holtzman wrote to Alzforum in an email. “It will be important in future experiments to increase or decrease expression of human ApoE in mice that only express human ApoE in the absence of murine ApoE to best interpret what one would see in humans.”
Mary Jo LaDu and Leon Tai, University of Illinois at Chicago, commented that the breadth of techniques used in this paper paint a comprehensive picture of how Aβ and plaque pathology relate to ApoE isoforms. LaDu was particularly intrigued by the clear impact ApoE2 had on reversing plaque pathology. “People so often ignore ApoE2 because it’s rare and costly to study,” she told Alzforum. “However, I think we can learn a lot from ApoE2, and here’s the evidence. This type of complex study is critical for understanding the big picture.”
While complimenting the paper overall, Rada Koldamova, University of Pittsburgh, cautioned that the levels of ApoE protein in each of the mouse groups were not identical. Levels of ApoE2 exceeded those of ApoE3, which were higher than ApoE4. “Because of that, I’m not convinced that increasing the level of ApoE4 would be harmful,” she told Alzforum. It could be that drugs that raise levels of any form of human ApoE, including ApoE4, would help clear plaques. She recommended looking within the individual groups to see if levels of the protein correlate with plaque clearance.—Gwyneth Dickey Zakaib
References
News Citations
- St. Louis: ApoE—A Clearer View of its Role In AD?
- Upping Brain ApoE, Drug Treats Alzheimer's Mice
- Bexarotene Revisited: Improves Mouse Memory But No Effect on Plaques
Paper Citations
- Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM. Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer's disease. Neurobiol Dis. 2002 Apr;9(3):305-18. PubMed.
- Liu G, Martins I, Wemmie JA, Chiorini JA, Davidson BL. Functional correction of CNS phenotypes in a lysosomal storage disease model using adeno-associated virus type 4 vectors. J Neurosci. 2005 Oct 12;25(41):9321-7. PubMed.
Other Citations
External Citations
Further Reading
Papers
- Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H, Basak JM, Finn MB, Holtzman DM. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-β amyloidosis. J Neurosci. 2011 Dec 7;31(49):18007-12. PubMed.
- Demattos RB. Apolipoprotein E dose-dependent modulation of beta-amyloid deposition in a transgenic mouse model of Alzheimer's disease. J Mol Neurosci. 2004;23(3):255-62. PubMed.
- Dumanis SB, Tesoriero JA, Babus LW, Nguyen MT, Trotter JH, Ladu MJ, Weeber EJ, Turner RS, Xu B, Rebeck GW, Hoe HS. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci. 2009 Dec 2;29(48):15317-22. PubMed.
News
- Could Bexarotene Treat Parkinson’s Disease?
- A Genetic Approach to the ApoE4 Puzzle
- Controlling Blood Pressure May Lower Amyloid in ApoE4 Carriers
- ApoE Does Not Bind Aβ, Competes for Clearance
- Does Estrogen Put the Brakes on Aging in ApoE4-Positive Cells?
- Does ApoE4 Risk Begin in the Womb?
- Does ApoE4 Lower Brain Metabolism Independently of Aβ?
- San Francisco: Tweaking Brain ApoE Reduces Aβ, Symptoms
- Upping Brain ApoE, Drug Treats Alzheimer's Mice
- Lowering ApoE Brings Down Amyloid in Mice
Primary Papers
- Hudry E, Dashkoff J, Roe AD, Takeda S, Koffie RM, Hashimoto T, Scheel M, Spires-Jones T, Arbel-Ornath M, Betensky R, Davidson BL, Hyman BT. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci Transl Med. 2013 Nov 20;5(212):212ra161. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Mayo Clinic
In this elegantly performed study, Hudry and colleagues addressed a few important gaps in the literature on ApoE and its links to amyloid pathology and Alzheimer’s disease. By using a still-unexplored strategy of adding the three different human ApoE isoforms to AD mouse models using gene therapy, and doing so at ages when the animals have existing amyloid pathology, the authors showed a detrimental effect of ApoE4, whereas ApoE2 appeared beneficial. In light of earlier reports, the current findings could have been expected, and they confirm that ApoE4 promotes Aβ pathology, even when added to the brain when amyloid is present. Importantly, that ApoE2 reduced amyloid burden in the same system suggests it could have pharmaceutical potential, and that needs to be further explored. The authors also report some ApoE isoform-dependent differences in ApoE concentrations amongst the mice. These findings are in line with previous results (see Riddell et al., 2008).
Caution should be used when interpreting these data. APOE genotype-dependent differences in ApoE concentrations have repeatedly been described in humans; however, the results have been inconsistent. Affinity differences between ApoE antibodies and the different ApoE isoforms most probably explain these reported inconsistencies. For instance, ApoE data from a WUE4-based ELISA (Wahrle et al., 2007), similar to the one Hudry et al. used, correlated poorly (r2=0.22) with those obtained using mass-spectrometry (Cruchaga et al., 2012). Further, Hudry and colleagues do not specify whether the samples from mice with different ApoE isoforms were analyzed with the same recombinant ApoE isoform as standard.
If the observed differences hold true, however, another important question arises: Could the observed effects be due partly to concentration-dependent effects of ApoE? For instance, would an increase in ApoE4 to the levels matching the reported ApoE2 levels have the same effect on amyloid plaque burden? Also, the authors mention a possible interaction between rodent ApoE and human ApoE, possibly in an isoform-dependent manner, which may bias the results. Lastly, whether the observed effects on amyloid plaque burden have any effect on cognitive function in these two mouse models needs to be established. Future studies addressing these questions are crucial for an adequate interpretation, not only of Hudry and colleagues’ results, but also of various similar studies aiming at elucidating the functional link between ApoE and AD.
References:
Riddell DR, Zhou H, Atchison K, Warwick HK, Atkinson PJ, Jefferson J, Xu L, Aschmies S, Kirksey Y, Hu Y, Wagner E, Parratt A, Xu J, Li Z, Zaleska MM, Jacobsen JS, Pangalos MN, Reinhart PH. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J Neurosci. 2008 Nov 5;28(45):11445-53. PubMed.
Wahrle SE, Shah AR, Fagan AM, Smemo S, Kauwe JS, Grupe A, Hinrichs A, Mayo K, Jiang H, Thal LJ, Goate AM, Holtzman DM. Apolipoprotein E levels in cerebrospinal fluid and the effects of ABCA1 polymorphisms. Mol Neurodegener. 2007;2:7. PubMed.
Cruchaga C, Kauwe JS, Nowotny P, Bales K, Pickering EH, Mayo K, Bertelsen S, Hinrichs A, Alzheimer's Disease Neuroimaging Initiative, Fagan AM, Holtzman DM, Morris JC, Goate AM. Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer's disease. Hum Mol Genet. 2012 Oct 15;21(20):4558-71. Epub 2012 Jul 20 PubMed.
View all comments by Henrietta NielsenUniversity of British Columbia
Although the association of ApoE with AD risk and age of onset is indisputable, whether ApoE is a tractable therapeutic target remains an open question. Brad Hyman’s group has recently published a paper that adds considerable strength to the therapeutic potential of the protective ApoE2 isoform. In this work, they used intraventricular injection of adeno-associated virus to deliver human ApoE2, ApoE3, or ApoE4 to the brains of symptomatic 7-month-old APP/PS1 transgenic mice. Two months after injection, they observed transduced cells in the choroid plexus and ependyma, with human ApoE levels constituting approximately 10 percent of endogenous murine ApoE levels. Five months after injection, the levels of human ApoE stabilized to be approximately 5 percent of endogenous murine ApoE. Two methods were used to determine if transduced human ApoE was distributed throughout the brain. First, species-specific immunofluorescence microscopy confirmed detection of human ApoE around amyloid deposits in transduced APP/PS1 mice. Second, microdialysis of interstitial fluid (ISF) confirmed the presence of ApoE when delivered into ApoE-/- mice, albeit in these animals the stable levels of transduced ApoE was approximately twice as much as in transduced APP/PS1 mice.
Five months after injection, amyloid plaque burden was unchanged in animals injected with human ApoE3, significantly increased in animals injected with ApoE4 and significantly decreased in animals injected with ApoE2. Insoluble and soluble Aβ40 and Aβ42 levels also demonstrated this ApoE4>ApoE3>ApoE2 pattern. By two months after injection, similar trends were observed, suggesting that ApoE’s effect on Aβ metabolism developed over a period of several months. No changes were observed in APP processing, glial activation, or expression of the Aβ peptidase insulin-degrading enzyme. Injection of ApoE2 increased plasma Aβ40 levels compared with ApoE3 and ApoE4, consistent with a role for ApoE in retaining Aβ within the CNS. No marked changes in blood brain barrier (BBB) integrity were observed. The rate of amyloid formation and clearance was evaluated using in-vivo, two-photon imaging, which showed that the rate of amyloidosis was greater in the presence of ApoE4 but significantly slowed in the presence of ApoE2 relative to ApoE3.
Array tomography was then used to investigate the effects of human ApoE expression on synaptic integrity, as ApoE4 is associated with higher levels of synaptic oligomeric Aβ and lower synaptic density in human AD brain. Transduction of ApoE3 or ApoE4 both led to loss of presynaptic synapsin-1 near amyloid plaques, an effect that was not observed with ApoE2. Compared with ApoE2 and ApoE3, ApoE4 led to increased dystrophic neurites and a significant loss of postsynaptic PSD-95 levels, again only in the vicinity of plaques.
To validate these findings in a second AD mouse model, symptomatic Tg2576 mice were transduced between 16 and 18 months of age and assessed three months afterwards. Microdialysis showed that ApoE4 led to significantly higher levels of oligomeric Aβ compared with ApoE2 or ApoE3 in ISF and significantly increased formic acid-extracted Aβ42.
In 2005, Steven Paul’s laboratory performed similar studies using lentiviral-mediated delivery of human ApoE directly into the hippocampus of PDAPP mice (Dodart et al., 2005). In these experiments, significantly elevated plaque and insoluble Aβ42 levels were observed five weeks after ApoE4 was injected into the CA3 region of 11- to 13-month-old PDAPP mice lacking endogenous murine ApoE. In contrast, neither plaque nor Aβ42 loads were altered in animals injected with ApoE2 or ApoE3. However, ApoE2 did decrease Aβ42 and plaque loads in a second group of PDAPP mice that were injected in the CA1 region at 7 months and assessed five weeks later. In this group of mice, ApoE4 tended to increase plaque burden, but the effects were not significant. However, pooling the findings of both groups revealed a significant reduction in hippocampal Aβ burden with ApoE2 and a significant increase with ApoE4 compared with lenti-GFP controls. A third group of PDAPP mice were injected at 10 months of age and examined three months later to assess the chronic effects of lentiviral delivery of ApoE. In this group of animals, ApoE2 significantly reduced hippocampal Aβ burden, but the increase in Aβ burden in ApoE4-injected animals did not reach significance. The authors also noted that lentivirus exposure led to loss of granule neurons in the dentate gyrus in animals examined three months after injection, whereas this neurotoxicity was not observed five weeks after injection. Neurotoxicity appeared to be confined to animals injected into the CA3 region, as no granule cell loss was reported after CA1 delivery.
Building on this foundation, the data provided by Hurdy et al. provide several new findings that support the feasibility of ApoE gene therapy approaches for AD. First, intraventricular AAV injection leads to stable transduction of the choroid plexus and ependymal cells, which leads to widespread distribution of transduced ApoE, including its presence in ISF. Second, even modest expression—namely 10 percent—of endogenous murine ApoE significantly accelerated Aβ clearance with ApoE2>ApoE3>ApoE4 in symptomatic animals. Third, these changes in Aβ metabolism corresponded with preservation of synaptic integrity in the vicinity of plaques. Finally, AAV delivery appeared to cause no overt neurotoxicity or BBB damage.
A pivotal future experiment will be to determine whether delivery of ApoE2 normalizes the deleterious effects of ApoE4 on Aβ metabolism and synaptic function. If so, AAV-mediated gene therapy of ApoE2 for AD could be pursued as a therapeutic approach for AD. AAV offers several advantages as a gene therapy vector and more than 60 clinical trials using AAV have been conducted in humans. In 2012, alipogene tiparvovec became the first AAV-mediated gene therapy to be approved for lipoprotein lipase deficiency in the European Union (European Medicines Agency, July 19, 2012). The advantages of AAV for gene therapy include the wide array of available serotypes and stable expression without integration of the vector into the host genome. Although its cargo capacity is relatively small compared with other vectors, ApoE is within its packaging limit. Importantly, direct CNS delivery may reduce some of the concerns regarding limitations of the host immune response for systemic delivery approaches. For example, Parkinson’s disease patients treated with bilateral intrastriatal infusion of AAV expressing human aromatic l-amino acid decarboxylase (AADC) exhibited improved mean scores on the Unified Parkinson Disease Rating Scale (UPDRS) by approximately 30 percent, although the surgical procedure led to an increased risk of intracranial hemorrhage and headache (Christine et al., 2009). A follow-up study that followed subjects annually for four years confirmed stable expression of AADC over this time period with an acceptable safety profile, albeit with slowly deteriorating UPDRS measures.
Strategies to overcome the deleterious effects of ApoE4 are urgently needed for millions of people who are affected by or at risk for AD. Hurdy and colleagues have taken the foundation laid by Dodart et al. a major step forward with this endeavor.
References:
Dodart JC, Marr RA, Koistinaho M, Gregersen BM, Malkani S, Verma IM, Paul SM. Gene delivery of human apolipoprotein E alters brain Abeta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2005 Jan 25;102(4):1211-6. PubMed.
Christine CW, Starr PA, Larson PS, Eberling JL, Jagust WJ, Hawkins RA, Vanbrocklin HF, Wright JF, Bankiewicz KS, Aminoff MJ. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology. 2009 Nov 17;73(20):1662-9. PubMed.
View all comments by Cheryl WellingtonUniversity of Southern California
In this interesting study, the authors demonstrate that the ApoE4 gene variant increases amyloid plaque load, promotes synaptic loss and neuritic dystrophy in close proximity to the plaques, and delays Aβ clearance. In contrast, ApoE2 expression decreases amyloid plaque density and soluble Aβ peptides in the brain, while the synaptic loss near the plaques is not increased compared to controls (AAV-GFP animals). These data support previous studies showing that ApoE4 causes extensive Aβ deposition by potentially delaying Aβ peptide clearance, leads to blood brain barrier disruption by activating the proinflammatory cyclophilin A pathway in BBB-associated pericytes, and is associated with the severity of cerebral amyloid angiopathy (CAA). This study offers important new insights into the effects of ApoE and suggests that ApoE2 may demonstrate neuroprotective properties. However, ApoE2 over-expression has been also implicated in CAA and it has been reported that ApoE2 is a risk factor for hemorrhage, while other studies connect ApoE2 with increased risk for atherosclerosis. In addition, it would be helpful to demonstrate whether the effects of ApoE2 are beneficial for learning and memory. More studies are needed to address these important questions.
View all comments by Angeliki NikolakopoulouWashington University
This paper beautifully shows that by using a unique method of AAV-mediated gene delivery via the choroid plexus and ependyma, relatively small increases in expression of human ApoE4 are achieved that increase Aβ deposition and toxicity, while ApoE2 has the opposite effect. Consistent with previous work from our lab and the Zlokovic lab, the data are consistent with ApoE3 and ApoE4 causing relative retention of Aβ within the brain. These data are extremely important because they suggest that increasing expression of ApoE2 may be protective and that decreasing expression of ApoE3 and ApoE4 may be protective in regard to Aβ deposition and Aβ toxicity. This is consistent with experiments published by my lab and the lab of Yadong Huang showing that decreasing human ApoE3 or ApoE4 from birth decreases Aβ deposition. Importantly, the experiments by Hudry et al. were done in adult animal animals, suggesting that if manipulations like this can be done in the adult brain, one would want to increase ApoE2 and decrease ApoE4 for therapeutic efficacy. It will be important in future experiments to increase or decrease expression of human ApoE in mice that only express human ApoE in the absence of murine ApoE to best interpret what one would see in humans.
View all comments by David HoltzmanVoyager Therapeutics
The paper by Hudry et al. is a beautiful extension of a paper my lab published almost nine years ago using viral vectors to deliver ApoE isoforms to the brains of PDAPP mice (see Dodart et al., 2005). In our study we showed that direct intrahippocampal administration of ApoE4 (using a lentiviral vector) rapidly increased Aβ/amyloid plaque burden, and by contrast administration of ApoE2 had quite the opposite effect, dramatically reducing hippocampal Aβ/amyloid burden, observed only five weeks after administration. In our experiments, the amount of each human ApoE isoform expressed was virtually identical, and thus we concluded that there was a marked qualitative/bidirectional effect/difference in the impact of these two ApoE isoforms on amyloid pathology. Remarkably, "gene delivery" of ApoE2 rapidly reduced brain amyloid burden against a background of mouse ApoE, which we had earlier shown was highly amyloidogenic in this (and other) mutant APP mouse models of AD (see Bales et al., 1997). As an aside, the effect of viral-mediated expression of ApoE2 in our PDAPP mouse model was more rapid and robust than observed with many/most anti-Aβ antibodies. We proposed in our paper that gene delivery of ApoE2 to the CNS could constitute a therapeutic approach to preventing or treating AD. This recent work by Hudry et al. and additional work from our laboratory (see Zhao et al., 2013 Society for Neuroscience meeting abstract) further support this strategy.
Recent advances in gene delivery using AAV vectors suggest that specific AAV serotypes and less-invasive routes of administration (e.g., intraventricular or intrathecal) may result in broad expression of ApoE2 in affected or vulnerable brain regions, sufficient perhaps to mitigate whatever pathogenic process underlies the clear "protective" effect of ApoE2. One cautionary note (among several) is whether broad delivery can be achieved in a much larger brain via these relatively noninvasive routes. Studies in nonhuman primates are underway to address this question and to assess safety ... but perhaps such a strategy is indeed plausible, as we postulated back in 2005.
References:
Dodart JC, Marr RA, Koistinaho M, Gregersen BM, Malkani S, Verma IM, Paul SM. Gene delivery of human apolipoprotein E alters brain Abeta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2005 Jan 25;102(4):1211-6. PubMed.
Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, Piccardo P, Ghetti B, Paul SM. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997 Nov;17(3):263-4. PubMed.
The Feinstein Institute for Medical Research
North Shore LIJ Hofstra SoM
We read with interest Hudry and Hyman's comprehensive set of studies examining the effects of ApoE2 and E4 isoforms in transgenic mice. Our own recent work, now in press at Molecular Psychiatry and all in humans (ex vivo and in vivo), nicely complements their work and replicates with fidelity earlier data in APOE transgenic models of Steve Paul and colleagues. In postmortem cortex we identified several mechanisms by which ApoE2 might confer neuroprotection: 1. Protein levels of ApoE were significantly greater in the brains of E2/E3 carriers than E3/E3 or, strikingly, E4 carriers; 2. By microarray E2 was associated with decreases in LTP-related transcripts and increases in extra-cellular matrix/integrin transcripts; 3. Downregulation in CypA, a molecule thought to be important for BBB integrity. Neuroprotection was also borne out in healthy elderly APOE2 controls, who demonstrated an "anti-AD profile" in CSF biomarkers, namely high levels of Abeta and low levels of t-tau and p-tau. In our view, abundance of E2, coupled to its well-known inability to bind to the LDLR receptor, may improve its ability to clear Abeta either through cells or at the BBB. Additional E2-associated reductions in LTP might also reduce activity-dependent secretion of Abeta.
References:
Conejero-Goldberg C, Gomar JJ, Bobes-Bascaran T, Hyde TM, Kleinman JE, Herman MM, Chen S, Davies P, Goldberg TE. APOE2 enhances neuroprotection against Alzheimer's disease through multiple molecular mechanisms. Mol Psychiatry. 2014 Feb 4; PubMed.
Make a Comment
To make a comment you must login or register.