Toronto: Human Amyloid Imaging Conference Showcases a Maturing Field
Quick Links
Call it development on fast-forward. Since the first major paper on human amyloid imaging appeared in the scientific literature (Klunk et al., 2004), a burgeoning field of research has sprung up and a recent conference illustrated how it has sped from infancy to adolescence in six years. Sensing an opportunity, multiple diagnostic imaging and pharma companies got into the act even as academic researchers the world over adopted this nascent technique. At this point, researchers are exploring myriad scientific questions about how to visualize the amyloid pathology in a living person’s brain and how to interpret what they see. For their part, commercial players are racing to satisfy FDA requirements for market approval of a slew of candidate ligands. The 4th annual Human Amyloid Imaging (HAI) Conference, held on 8 April 2010 in Toronto, Canada, showcased this dual message clearly. Organized largely by Keith Johnson of Massachusetts General Hospital, with help from Bill Jagust of University of California, Berkeley, and Bill Klunk and Chet Mathis of the University of Pittsburgh Medical School, this meeting drew some 160 attendees from academia and industry worldwide for a day of short talks, two keynote lectures, and the ample, freewheeling discussion that is a mark of distinction for this conference.
One the one hand, there seemed to be no doubt in the room that amyloid imaging works. That is, the main ligands in use—six at present by this writer’s count—all reliably and reasonably specifically image β amyloid deposits in the brain. There was a shared sense that four different commercial F18-labeled ligands, each of which is at a different point in the clinical development pipeline, overall appear to perform quite similarly. And in a sign that the field is beginning to mature, the how-to debate has shifted to smaller methodological issues. Investigators now see the need to work out a degree of standardization on the fine points of data acquisition and analysis so they can better compare findings from study to study. The twin excitement here lies in readying amyloid imaging for robust, larger-scale application in clinical trials as well as to support an earlier, biomarker-driven diagnosis of Alzheimer disease.
On the other hand, the use of these amyloid imaging agents for scientific exploration of the aging brain’s underlying biology has merely begun to scratch the surface. Here, the excitement lies in peeling back a deeper layer of the brain’s mysteries and discovering something fundamentally new. Scientists are increasingly combining amyloid imaging with other forms of measurement in cognitively normal people, such as paper-and-pencil tests, several different modes of brain imaging, AD risk genes, even indicators of cardiovascular health such as blood pressure and vascular amyloid. They do this to address the question of what makes some aging people succumb to the presence of β amyloid in their brains while others can “live at peace with their amyloid for many years,” as Sperling put it. Scientists are hoping that this effort will eventually explain the pathophysiology of AD. On this front, scientists agreed, the field is only just getting underway. Hence, it is at present producing tantalizing but discrepant results that need resolution.
A presentation on this topic won the HAI conference’s $500 Young Investigator Award. Alex Becker, a research fellow at Massachusetts General Hospital, Boston, took a crack at prying apart the factors that together might determine how well aging people can tolerate amyloid accumulating in their brains, and how metabolism is affected by increasing levels of amyloid. Becker measured FDG metabolism, an indicator of how much glucose the brain uses (i.e., how active it is or how hard it is working). He did so in 77 cognitively normal people and focused in on the 21 among them who were amyloid positive. As a group, their metabolism was reduced compared to the amyloid-negative group in cortical areas that form the default mode network, but Becker saw a lot of individual variability. Clearly, other factors were playing a role. To unmask those, Becker first looked at ApoE status. He found that the ApoE4 carriers exhibited a steeper decline in FDG metabolism with advancing age than did the non-carriers. This was region-dependent and primarily the case in frontal regions. Intriguingly, younger ApoE4 carriers started out with higher FDG uptake in certain brain regions than older carriers, even if both had equal amounts of amyloid, and then declined faster.
If ApoE accounted for only a part of the scatter in the initial data, what else was going on? Becker also studied cognitive reserve. He did so in two ways, expressed by way of education and of AMNART scores. (The AMNART is an intelligence test that was designed to be somewhat resistant to the effects of aging.) In this analysis, FDG metabolism went up along with intelligence scores and education in amyloid-negative people, as might be expected. But in amyloid-positive people, Becker found the opposite: FDG metabolism was lower in people with high intelligence/more education. “That is why we did not see this in the pooled group,” Becker told the audience, referring to the lack of a significant relationship between FDG and AMNART in the full group versus the split group. This might suggest that people with high cognitive reserve remain outwardly cognitively normal even though their brain metabolism is already declining.
Along similar lines, Alexander Drzezga of the Technical University in Munich, Germany, reported on his fMRI study of resting-state connectivity in 12 PIB-negative and 12 PIB-positive cognitively normal people as well as 13 PIB-positive people with mild cognitive impairment (MCI). This study detected early signs that the connections between cortical “hubs” in the brain are already beginning to break down in cognitively normal people who were amyloid-positive. This fMRI connectivity loss overlapped with the reduced glucose metabolism that Becker had analyzed. Reisa Sperling of Brigham and Women’s Hospital, Boston, added a cognitive testing angle to this emerging story. Her group’s new analysis of the normal control group in the florbetapir Phase 2 trial suggests that even within what is considered the normal range of an episodic memory test, a higher level of amyloid deposition was associated with lower performance. Overall, the talks generated the impression that amyloid imaging in cognitively normal people is beginning to correlate well with other imaging modalities, and that a larger picture is emerging of subtle deficits across a broad range of indicators in outwardly normal people who have amyloid in their brains. The scientists agreed that more research in larger samples is needed to fill in this emerging picture.
Beyond genetics and cognitive reserve, Vladimir Hachinski, a stroke expert at University of Western Ontario, London, Canada, pleaded with his amyloid imaging colleagues to take vascular disease into consideration. Instead of being excluded from AD studies, people with vascular disease should be made a focus of study because “when you have vascular disease, that is when amyloid causes cognitive impairment,” Hachinski said (see also MCI conference). This view drew widespread agreement. (Interested to learn more? See upcoming Cerebral Amyloid Angiopathy Conference.) Likewise, the day heard repeated calls for imaging inflammation as a modifying factor, though no one appeared to be aware of suitable ligands beyond 11C PK11195, which has not found widespread acceptance.
The HAI Conference saw intense discussion about the technical challenges amyloid imaging is confronting at present. They seemed solvable. Above all, scientists need to agree on which brain area to use as a normalization region (see Part 2 of this series) and on how to draw its contours, so that studies at different sites become more comparable. Scientists shared their troubles with a technical problem called partial volume effects, and discussed whether to try to tame it with what’s called a CSF correction. The correction measures more amyloid in atrophic brains but less so in non-atrophic brains. Many scientists agreed that it is best to process the data with and without this correction and evaluate what the correction does to the outcome.
Speakers and audience members also argued about which analysis methods work best for particular goals, i.e., multicenter drug studies versus academic explorations of exactly where an amyloid scan becomes “positive” along a poorly understood process of accumulation. They considered different ways of setting cutoffs for this transition. They agonized over how they can much more rigorously define what it means that someone is “cognitively normal.” This might lessen a selection bias they suspect of being at the root of discordant results in current research of cognitively normal people, which are causing some confusion at this early stage of research. “Nobody goes to bed on Monday and wakes up impaired on Tuesday,” said Cliff Jack of the Mayo Clinic in Rochester, Minnesota. “It is a matter of where on the continuum you draw your sample. Most samples are small, most have biases, and that explains the differences between the studies out there.” (For more on these issues, see Part 2 of this series.)
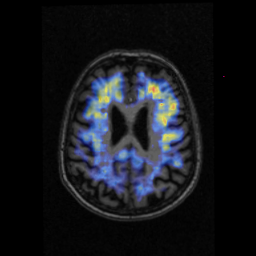
PIB-PET From a Clinically Normal 80-Year-Old Woman
Her mean cortical PIB is below threshold, but she has focal deposition (yellow-red). Many normal subjects have clear focal uptake but have average cortical uptake that is insufficient to categorize them as amyloid-positive. Longitudinal studies will tell if this uptake accumulates over time. Image credit: Keith Johnson, Massachusetts General Hospital
In contrast, one finding stood out for its clarity throughout the day. With great consistency, the ApoE4 AD risk allele shows up in the AD-prone group of very mildly impaired or cognitively normal volunteers who have amyloid. Mark Mintun of Washington University, St Louis, Missouri, showed how ApoE4 carriers trend upwards in their amyloid load at younger ages than non-carriers; Elizabeth Mormino of the University of California, Berkeley, found ApoE4 carriers overrepresented among the amyloid-positive subgroup in her study of cognitively normal volunteers. Kenji Ishii of the Tokyo Metropolitan Institute of Gerontology, Japan, found this link in the J-ADNI cohort, as well; there, all ApoE4-carrying AD and MCI patients to date were amyloid-positive, as were half the ApoE4-carrying cognitively normal participants. Osama Sabri of the University of Leipzig, Germany, presented data on a link between carrying an ApoE4 allele and having brain amyloid as measured by the ligand florbetaben; in this study, uptake of the amyloid ligand went up with each copy of the allele in AD patients.
The poster session deepened this impression. Christopher van Dyck at Yale University School of Medicine in New Haven, Connecticut, tested in his group’s own study population the previous finding that ApoE4 drives preclinical amyloid deposition in a dose-dependent way (Reiman et al., 2009). The Yale investigators ran PIB-PET scans in 270 cognitively normal volunteers in their fifties and sixties who had a first-degree relative with Alzheimer’s, and confirmed that in their sample, too, ApoE4 carriers had considerably more amyloid than non-carriers of the same age, sex, and education. Amyloid-positive volunteers tended to be slightly weaker on tests of episodic memory. Shizuo Hatashita of Shonan Hospital in Atsugi, Japan, reported that among 34 people with MCI who received a PIB scan and were followed clinically for up to two years, the PIB-positive ApoE4 carriers progressed faster to meet an AD diagnosis.
These new data jibe with recent published reports suggesting that ApoE4 carriers tend to accumulate more brain amyloid than non-carriers (e.g., Drzezga et al., 2009), do so at younger ages (Morris et al., 2010), age with reduced blood flow in the brain (Thambisetty et al., 2010), and have lower CSF Aβ42 levels in various biomarker studies including ADNI. “The ApoE effect on amyloid deposition seems incredibly concordant,” observed Neil Buckholtz of the National Institute on Aging. Buckholtz qualified, however, as did other scientists, that this seemingly definitive conclusion might yet shift if independent research confirms a paper claiming that some of the ApoE effect on age of disease onset is actually the doing of the nearby gene TOMM40 (Roses et al., 2009; Lutz et al., 2010).
Beyond implicating ApoE4 in amyloid deposition, researchers at HAI exchanged news on various fronts. For example, Ishii presented the first amyloid imaging results from the Japanese ADNI (J-ADNI) study. In brief, J-ADNI is going well, Ishii said, having enrolled 354 out of the desired 600 participants as of this month. Thirteen amyloid imaging sites to date have imaged 21 people with AD, 28 with MCI, and 46 controls aged 66 to 74. Of those, 95, 75, and 24 percent, respectively, have proven amyloid-positive. Overall, J-ADNI’s visual assessment performed as well as the quantitative one, but small amounts of amyloid in borderline positive cases are better detected with dynamic data acquisition and DVR (see Part 2), Ishii said.
First data on epidemiological modeling of brain amyloid in the elderly population were on the menu (see Part 2), as well as Phase 3 autopsy validation for the 18F ligand florbetapir (see Part 3). Also new were Phase 2 data on two other 18F ligands (see Part 4). First human data on a fourth ligand, which particularly intrigued some imagers, were presented at the Springfield Conference in Geneva last month. To learn all about that, see Part 5.—Gabrielle Strobel.
This is Part 1 of a six-part series. View a PDF of Part 1. See also Part 2, Part 3, Part 4, Part 5, Part 6. View PDF of the entire series.
References
News Citations
- Toronto: Ah, The Devil in the Details
- Toronto: Last Gift to Science—Hospice Patients Validate Amyloid Ligand
- Toronto: Sister 18F Ligands Jostle for Primacy
- Geneva: The AstraZeneca Ligand—The Fairest of Them All?
- Toronto: HAI Amyloid Imaging Conference Abstracts
Paper Citations
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, Ausén B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Långström B. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. 2004 Mar;55(3):306-19. PubMed.
- Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, Ayutyanont N, Keppler J, Reeder SA, Langbaum JB, Alexander GE, Klunk WE, Mathis CA, Price JC, Aizenstein HJ, Dekosky ST, Caselli RJ. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc Natl Acad Sci U S A. 2009 Apr 21;106(16):6820-5. PubMed.
- Drzezga A, Grimmer T, Henriksen G, Mühlau M, Perneczky R, Miederer I, Praus C, Sorg C, Wohlschläger A, Riemenschneider M, Wester HJ, Foerstl H, Schwaiger M, Kurz A. Effect of APOE genotype on amyloid plaque load and gray matter volume in Alzheimer disease. Neurology. 2009 Apr 28;72(17):1487-94. PubMed.
- Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, Mintun MA. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010 Jan;67(1):122-31. PubMed.
- Thambisetty M, Beason-Held L, An Y, Kraut MA, Resnick SM. APOE epsilon4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch Neurol. 2010 Jan;67(1):93-8. PubMed.
- Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, Huentelman MJ, Welsh-Bohmer KA, Reiman EM. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer's disease. Pharmacogenomics J. 2010 Oct;10(5):375-84. Epub 2009 Dec 22 PubMed.
- Lutz MW, Crenshaw DG, Saunders AM, Roses AD. Genetic variation at a single locus and age of onset for Alzheimer's disease. Alzheimers Dement. 2010 Mar;6(2):125-31. PubMed.
Other Citations
External Citations
Further Reading
News
- PIB-PET Biomarker Study Confirms Bapineuzumab Lowers Amyloid
- Multi-Paper Alert: More Data That Brain Amyloid Is Bad for You
- HAI Seattle: Does Brain Amyloid Correlate With Sagging Metabolism?
- Cortical Hubs Found Capped With Amyloid
- Imaging Studies Support Cognitive Reserve Theory
- Phase 2 Trial of Amyloid Tracer for AD Is Launched
- Toronto: HAI Amyloid Imaging Conference Abstracts
- St. Louis: Imaging Preclinical AD—Can You See it Coming in the Brain?
- BOLD New Look—Aβ Linked to Default Network Dysfunction
- HAI Seattle: Aβ May Disrupt Brain Function in Normal Seniors
- HAI Seattle: Not Just Amyloid, Not Just PIB
- HAI Seattle: Biomarkers Closing in on AD Pathological Sequence?
- Paper Alert: PIB-PET, Brain Biopsies Match Well for Detecting Aβ
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.