St. Louis: Imaging Preclinical AD—Can You See it Coming in the Brain?
Quick Links
At the 7th Leonard Berg Symposium, brain imaging researchers of various stripes—some MRI, some FDG PET, some amyloid imaging aficionados—took stock of what brain imaging tells the field about the preclinical period of Alzheimer disease. From their vantage point, a picture is slowly coming into focus whereby a number of different imaging techniques together predict that a person is on the way to developing Alzheimer’s symptoms. For some speakers, the evidence is strong enough to start thinking seriously about how to use images (in conjunction with CSF and cognition) to test prevention of AD in people who are at high risk, either by virtue of having inherited the ApoE4 risk allele, or a dominant mutation.
Nick Fox of King’s College, London, UK, reminded the audience that of the imaging modalities currently used, magnetic resonance imaging (MRI) is one that can yield perhaps the most exquisite view of the brain with a resolution of 1 mm. This can be had with a 10-minute scan session that is robust and practical for the imaging technician and easy to tolerate for the subject, certainly for presymptomatic people and even for many patients. This method is particularly suited for tracking change, for example, shrinking of brain areas, over time, and to measure progression. “We are all interested in drugs that slow or stop progression,” Fox said.
Some prior imaging studies on presymptomatic eFAD mutation carriers have been done, all very small (Ridha et al., 2006; Godbolt et al., 2006; Ringman et al., 2007; Ginestroni et al., 2009). Early data from Fox’s group suggest that, after diagnosis, people with eFAD lose volume rapidly. Fox showed images of one person’s hippocampus shrinking dramatically within only two years of diagnosis. Many years prior to diagnosis atrophy rates are slow, but five to two years prior, they noticeably pick up to as much as 3 percent per year in particularly vulnerable areas. Rates come in plural form because they differ by brain area; for example, the entorhinal cortex shrinks particularly fast just before diagnosis. “Serial imaging gives us fantastic insight into structural change prior to diagnosis. The excitement of DIAN is that we can pull more people together to expand on these findings with better power,” Fox said.
Technically, the large range of normal variation in brain size between one person and the next makes it important to focus on rates of change in given areas, not absolute measures, and also to focus on the area that changes between two well-registered images (aka the boundary shift interval), without including the volume of the entire brain, Fox noted.
MRI is one of the most established ways of brain imaging. In the past five years, it got increasingly prominent company from amyloid positron emission tomography (PET), which visualizes in fiery color and in living people pathology that previously could only be seen after a patient had died. Research groups around the globe quickly adopted the amyloid label Pittsburgh Compound B (PIB) or similar compounds for study, and as people began to drill deeper, the story became more complicated. Questions arose about what it means that a large fraction of cognitively healthy elderly have amyloid in their brains, about exactly which forms of amyloid PIB binds to, and how being PIB positive related to other liabilities ascribed to the preclinical stage of AD. At the Leonard Berg Symposium, Keith Johnson of Massachusetts General and Brigham and Women’s Hospitals in Boston addressed these questions with the data available to date.
Multiple studies show that, by and large, everyone with AD has high PIB retention in their brains, Johnson said. There are exceptions relating to the maturity of plaques and perhaps “strains” of Aβ (e.g., Rosen et al., 2009). “These are interesting and important, but they are exceptions,” Johnson stressed. At the early symptomatic MCI stage, people either have a control pattern or an AD-like pattern, not an in-between pattern. The so-called PIB-positive (PIB+) controls, that is, people who are clinically normal but have amyloid in their brains, tend to have it in a stereotyped AD-like distribution with a weaker intense signal. Around the time of diagnosis, PIB binding tends to become saturated and stay relatively stable through the course of clinical disease.
One burning question on the minds of people who study preclinical detection in different populations and with different methods is how the data might all fit together. Johnson offered this hypothesis:
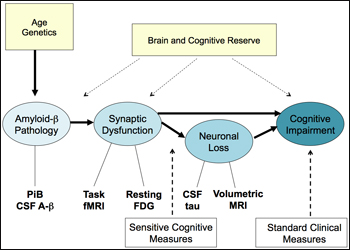
By that scenario, the long road to disease would begin with amyloid-β pathology. Age and genetics (i.e., ApoE genotype) influence a person’s propensity to deposit amyloid, and both CSF Aβ and PIB PET can detect it. This would lead to synaptic dysfunction, which impairs cognition directly, and also indirectly over time as the neurons bearing malfunctioning synapses slowly die. It is widely accepted that brain and cognitive reserve influence how long a person can withstand the damaging effects of the encroaching pathology. Bill Klunk, of the University of Pittsburgh Medical School in Pennsylvania, noted that vascular disease is another important environmental factor that acts at this level. The PIB+ clinically normal people currently draw intense interest among researchers. Are they the ones who are on this path to AD? Imaging methods from functional MRI to FDG glucose PET and volumetric MRI exist to visualize and quantify these stages. The standard clinical tests used routinely in diagnosis pick up the latest stage on this flow chart, but increasingly, new findings are converging around more sensitive cognitive measures to pick up earlier, subtler changes (see Part 3 and below).
One way of probing whether this suggested signature of AD holds true is to test whether amyloid deposition in clinically and cognitively normal people relates to the other components in this diagram. “We do this by looking at other measures of AD in PIB+ controls,” Johnson said. Here are four examples of doing so that Johnson presented in his talk:
- Cortical thickness: In MGH studies of cognitively normal PIB+ people, increasing amyloid is associated with a thinning cortex in the precuneus, anterior and posterior cingulate, and some temporo-parietal and medial frontal cortical areas. These areas have come to represent a network that represents functional connection. “These areas talk to each other,” Johnson said.
- Hippocampal volume: In the MGH/BWH studies, this measure does not seem to relate to amyloid deposition in the cognitively normal volunteers but does by mildly impaired stages of CDR 0.5 and 1. “This suggests to us that the cortex thins first and the hippocampus a bit later,” Johnson said.
- Brain connectivity: A recent study reported that the more highly linked an area is—in other words, if it is a hub of intense connectivity—the more amyloid it tends to have (Buckner et al., 2009). This month, the researchers reported an amyloid-related disruption of intrinsic connectivity among asymptomatic elderly (Hedden et al., 2009). “This suggests a disconnection where one area is unable to talk with other, previously connected areas anymore,” Johnson said. In July, the same researchers reported that using fMRI, they found that brain amyloid in non-demented people mapped to their inability to deactivate the default network when focusing on a task (Sperling et al., 2009).
- Cognitive function: It is becoming increasingly clear that amyloid deposition is tied to cognitive impairment. IQ and education are important here because people high on those measures are able to tolerate amyloid pathology longer. People with low IQ/education scores show a much stronger link between amyloid in their precuneus (a particularly early area) and how they perform on neuropsychological tests (Rentz et al., Annals of Neurology, in press). And preliminary analyses of longitudinal data showed a detectable decline in a delayed word recall test. People declined along with increasing precuneus PIB. These are subtle effects on difficult tests, Johnson noted; the first difference the MGH/BWH studies see in those tests is that people with PIB in precuneus lose the practice effect, i.e., they don't improve anymore with repeated testing. “We really don’t understand in biological terms what cognitive reserve is and how it preserves function,” Johnson noted.
The Society for Neuroscience conference, held 17-21 October 2009 in Chicago, featured even newer data along these lines. To quote but one example, Deepti Putcha, working with Reisa Sperling at Brigham and Women’s Hospital in Boston, reported that cognitively normal older people who performed poorly on a challenging memory test (RVALT) had hyperactivation of their hippocampus along with cortical thinning in AD-vulnerable areas of the medial temporal lobe. These data are fresh out of the scanner, and concomitant amyloid and connectivity imaging data on this new aging cohort still needs to be analyzed, Putcha said.
These and other emerging clues on how multiple preclinical markers link up together in the same people will hopefully bring into focus a predictive signature over the course of the next five years, as longitudinal studies under way at several institutions progress. But even now, the field has advanced far enough to start planning prevention research, argued Eric Reiman of the Banner Alzheimer's Institute in Phoenix, Arizona. Evaluating promising presymptomatic treatments in the general population takes too long and is too costly, but doing so in high-risk groups such as ApoE4 carriers and presymptomatic carriers of dominantly inherited Alzheimer disease (DIAD)/eFAD mutations has come entirely within reach, Reiman said. Adding to the other speakers’ presentations a brief summary of his group’s research, Reiman noted that he has followed ApoE- genotyped volunteers—that is, people at three defined levels of risk—with brain imaging for well over a decade. People with the ApoE4 allele have decreased glucose utilization and subtle metabolic changes already as young adults, predating even the earliest biochemical marker to date, i.e., CSF Aβ. These FDG PET abnormalities do not progress continuously from early adulthood, but they flag early on where the AD changes will develop later, Reiman said (Reiman et al., 2001; Caselli et al., 2004; Reiman et al., 2005; Reiman et al., 2009).
The wealth of imaging and biomarker data the field has accumulated to date provides a sufficient foundation for proof-of-concept trials of biomarker outcomes, not clinical outcomes, with several hundred subjects, Reiman said. He spoke forcefully for starting an era of prevention research. This will not be a quick achievement, but it is time to begin gathering drug/biomarker data that can open a new regulatory path to approval. Such trials would also show research volunteers who have allowed scientists to poke, question, and scan them for decades that the scientists, in turn, will use all that data toward something tangible that truly matters to the volunteers and their families.—Gabrielle Strobel.
This is Part 6 of a seven-part series on presymptomatic detection. See also Parts 1, 2, 3, 4, 5, and 7.
References
News Citations
- St. Louis: Cognition Pre-dementia—Like eFAD, Like LOAD?
- St. Louis: Scientists, Families Target Preclinical Detection, Trials
- St. Louis: The Family View—What Do Study Volunteers Want From DIAN?
- St. Louis: Biomarkers Pre-dementia—Like eFAD, Like LOAD?
- St. Louis: Is Rare Familial Alzheimer’s a Model for the Millions?
- St. Louis: An eFAD Prevention Trial—One Man’s View
Paper Citations
- Ridha BH, Barnes J, Bartlett JW, Godbolt A, Pepple T, Rossor MN, Fox NC. Tracking atrophy progression in familial Alzheimer's disease: a serial MRI study. Lancet Neurol. 2006 Oct;5(10):828-34. PubMed.
- Godbolt AK, Waldman AD, Macmanus DG, Schott JM, Frost C, Cipolotti L, Fox NC, Rossor MN. MRS shows abnormalities before symptoms in familial Alzheimer disease. Neurology. 2006 Mar 14;66(5):718-22. PubMed.
- Ringman JM, O'Neill J, Geschwind D, Medina L, Apostolova LG, Rodriguez Y, Schaffer B, Varpetian A, Tseng B, Ortiz F, Fitten J, Cummings JL, Bartzokis G. Diffusion tensor imaging in preclinical and presymptomatic carriers of familial Alzheimer's disease mutations. Brain. 2007 Jul;130(Pt 7):1767-76. PubMed.
- Ginestroni A, Battaglini M, Della Nave R, Moretti M, Tessa C, Giannelli M, Caffarra P, Nacmias B, Bessi V, Sorbi S, Bracco L, De Stefano N, Mascalchi M. Early structural changes in individuals at risk of familial Alzheimer's disease: a volumetry and magnetization transfer MR imaging study. J Neurol. 2009 Jun;256(6):925-32. PubMed.
- Rosen RF, Ciliax BJ, Wingo TS, Gearing M, Dooyema J, Lah JJ, Ghiso JA, Levine H, Walker LC. Deficient high-affinity binding of Pittsburgh compound B in a case of Alzheimer's disease. Acta Neuropathol. 2010 Feb;119(2):221-33. PubMed.
- Buckner RL, Sepulcre J, Talukdar T, Krienen FM, Liu H, Hedden T, Andrews-Hanna JR, Sperling RA, Johnson KA. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer's disease. J Neurosci. 2009 Feb 11;29(6):1860-73. PubMed.
- Hedden T, Van Dijk KR, Becker JA, Mehta A, Sperling RA, Johnson KA, Buckner RL. Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. J Neurosci. 2009 Oct 7;29(40):12686-94. PubMed.
- Sperling RA, Laviolette PS, O'keefe K, O'brien J, Rentz DM, Pihlajamaki M, Marshall G, Hyman BT, Selkoe DJ, Hedden T, Buckner RL, Becker JA, Johnson KA. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009 Jul 30;63(2):178-88. PubMed.
- Reiman EM, Caselli RJ, Chen K, Alexander GE, Bandy D, Frost J. Declining brain activity in cognitively normal apolipoprotein E epsilon 4 heterozygotes: A foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer's disease. Proc Natl Acad Sci U S A. 2001 Mar 13;98(6):3334-9. PubMed.
- Caselli RJ, Reiman EM, Osborne D, Hentz JG, Baxter LC, Hernandez JL, Alexander GG. Longitudinal changes in cognition and behavior in asymptomatic carriers of the APOE e4 allele. Neurology. 2004 Jun 8;62(11):1990-5. PubMed.
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Correlations between apolipoprotein E epsilon4 gene dose and brain-imaging measurements of regional hypometabolism. Proc Natl Acad Sci U S A. 2005 Jun 7;102(23):8299-302. PubMed.
- Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, Ayutyanont N, Keppler J, Reeder SA, Langbaum JB, Alexander GE, Klunk WE, Mathis CA, Price JC, Aizenstein HJ, Dekosky ST, Caselli RJ. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc Natl Acad Sci U S A. 2009 Apr 21;106(16):6820-5. PubMed.
Further Reading
News
- St. Louis: Scientists, Families Target Preclinical Detection, Trials
- St. Louis: The Family View—What Do Study Volunteers Want From DIAN?
- St. Louis: Cognition Pre-dementia—Like eFAD, Like LOAD?
- St. Louis: Biomarkers Pre-dementia—Like eFAD, Like LOAD?
- St. Louis: Is Rare Familial Alzheimer’s a Model for the Millions?
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.