SfN: Return of the Other—Tau Is Back, Part 1
Quick Links
This year’s 35th annual meeting of the Society for Neuroscience, held last week in Washington, D.C., left a visitor without a lead news story, such as a new gene or a new experimental therapy. But upon closer inspection, the conference did have groups of researchers share a common sense of excitement about trends afoot, and one such trend revolves around the comeback of the microbule-stabilizing protein tau.
Of course, tau was never really gone. Ever since Alois Alzheimer’s original description of neurofibrillary tangles, it has been clear that tau is a central component of AD. But when the discovery of autosomal-dominant mutations in APP and presenilins in the early 1990s gave researchers something to sink their teeth into, the field’s attention turned overwhelmingly toward the amyloid side of the disease. The discovery a few years later of tau mutations that cause frontotemporal dementia did spur renewed interest in this protein. But these tau mutations do not cause AD, and for years the molecular and cellular biology research that usually follows a major genetic discovery occurred on a separate track from work on Aβ. The twain rarely met, and lectures were routinely given on one protein without much mention of the other. The advent of new tau mouse models has begun to change this, however. At the SfN conference, the Aβ peptide and tau—though not plaques and tangles—were increasingly mentioned together in the same breath.
Beyond the mouse research alone, tau is enjoying a general resurgence of interest across technical fronts. Last week’s SfN conference featured 124 presentations involving tau and AD, almost twice the number of the year before and up from 47 in the year 2000. Aficionados of the field will notice that half the names of presenting researchers in this news series are from labs that formerly investigated mostly the amyloid side of AD. Fueling this renewed interest further is a growing consensus around Virginia Lee’s and John Trojanowski’s observation that tau is an important common pathway to a wide range of neurodegenerative diseases that initially present a wholly different picture to the clinician. Besides AD, this includes Parkinson disease, frontotemporal dementias, and the lysosomal storage disease Pick’s. A large proportion of the major paths to age-related neurodegeneration, each with a different beginning, eventually lead through tau, Frank LaFerla and Eckhard Mandelkow jointly emphasized with Lee at an SfN news conference.
Ironically, just as Alois Alzheimer’s groundbreaking observation is approaching its 100th birthday next year, it appears as if, for all its crystallizing insight, it may have kept investigators pursuing the wrong suspect for decades. One early implication of the new wave of tau research is that it’s not primarily the tangles that cause problems in AD, much as they are realizing that on the amyloid side, it’s not primarily the plaques, either. What follows below and in three cross-linked stories is a four-part summary of tau news highlights from the SfN conference. It is not comprehensive. As always, conventioneers are cordially invited to share their own recollections, comments, corrections. See also Part 2, Part 3, and Part 4.
Tau Can’t Swim Upstream
Frank LaFerla, of the University of California, Irvine, strengthened the emerging connection between amyloid-β and tau as part of his laboratory’s ongoing analysis of the triple transgenic mice expressing APP, PS-1, and tau mutations. A series of 10 presentations at the meeting addressed various aspects involving tau. The scientists’ first major conclusion was that Aβ acts upstream of tau. “When we manipulate Aβ, either genetically or pharmacologically, it has a direct impact on tau. When we augment tau, either genetically or pharmacologically, that has no effect on Aβ. We see a one-way arrow going from Aβ to tau. The arrow going back is absent or small,” LaFerla summarized at a press conference. The body of research suggesting that Aβ acts on tau further downstream in the pathogenic cascade is broadening. Other labs have reported similar evidence, and at the conference Italian researchers from the universities of Florence and Brescia showed data suggesting that the TgCRND8 mutant APP-transgenic mice made in David Westaway’s laboratory (Chishti et al., 2001) develop tau hyperphosphorylation by 7 months of age.
In one presentation by the LaFerla lab, Salvatore Oddo and Antonella Caccamo turned around their previous observation that removing Aβ reduced the tau burden in the triple transgenic mice and asked whether changes in tau could likewise influence the Aβ burden. Before addressing this question directly, Oddo first documented the time course of tau pathology and tau kinase activity in the triple transgenic mice. A panel of p-tau antibodies and Gallyas silver stain on brain sections indicated that tau phosphorylation appeared around 12 months and increased with age. Tau extractions then showed that phosphorylation at the AT100 site precedes tau aggregation, beginning as early as 6 months of age, but phosphorylation at the PHF-1 site sets in only after tau has begun aggregating. Among tau kinases, GSK3β activity showed an increase around 15 months of age; CDK5 levels did not change as the mice aged but the concentration of its activator p25 increased by 15 months, Oddo reported. Perhaps the most important finding from this study came from crossing the triple transgenic mice to another strain that overexpresses human tau sixfold relative to endogenous tau. These tau-laden crosses predictably had increased steady-state levels of tau and more tau phosphorylation, but all this did not change their amyloid pathology at all. Oddo et al. saw no differences in APP or C99 levels, Aβ40 or 42 levels, or amyloid deposits. This expands previous data by this group showing that injection of anti-tau antibodies did not affect amyloid pathology.
Another presentation from the LaFerla group brought up the Aβ-tau connection from a different angle. Kim Green reported that glucocorticoids—stress hormones that are thought to mediate part of the environmental component of a person’s disease risk—increased APP production, BACE activity, intraneuronal Aβ levels, and also hippocampal tau accumulation in the triple transgenic mice. However, glucocorticoid treatment did not increase tau accumulation in transgenic mice lacking APP, suggesting that this effect of stress hormones is a downstream consequence of its effect on APP. (It also implies that glucocorticoid drugs should be prescribed to the elderly with caution.)
LaFerla’s second major conclusion is that soluble forms of tau act importantly in pathogenesis before either tangles or plaques are fully in place, LaFerla noted. This finding agrees with data from two other tau mouse models (Andorfer et al., 2005; SantaCruz et al., 2005). These models differ in that one expresses wild-type human tau without mouse tau and the other expresses an on-off regulatable form of human mutant tau, and yet they both suggest that soluble tau, not the tangles, appears to do the early damage to cognition.
Swim to Platform: Without Tau, Aβ-Loaded Mice Do Fine
Genetically increasing tau may not feed back into amyloid pathology, but what happens in the opposite experiment, that is, decreasing tau in the face of rapid amyloid production? Lennart Mucke of the University of California, San Francisco, chose this approach to address how tau contributes to AD. The cell biology of tau is well-studied and suggests a number of candidate mechanims—Is it the tangles? Loss of microtubule stabilization? Axonal transport? Or the cell cycle?—but in vivo data are more scarce. To address this issue, Erik Roberson in Mucke’s lab bred an APP-transgenic mouse that has well-characterized cognitive deficits and amyloid pathology to a heterogyzous tau deletion mouse. Surprisingly, knocking tau levels in half strongly protected the offspring. The mice with rampant APP expression but little tau performed almost normally in the water maze. They escaped other behavioral abnormalities seen in the APP transgenics. Measures of synaptic function, such as calbindin levels, were almost normal in the tau-depleted mice. By contrast, reducing tau did not change the mice’s amyloid pathology, the presence of dystrophic neurites, or aberrant axonal sprouting, much like increasing tau did not change amyloid pathology in Oddo and LaFerla’s experiment.
This experiment suggests yet again that cognitive performance in mouse models is separate from plaque pathology. Yet the most surprising conclusion from this work may be that the presence of tau—but not of tangles—is necessary for soluble forms of Aβ to wreak the previously noted synaptic and behavioral damage. The implication would be that soluble tau acts downstream of soluble Aβ in a pathway that attacks the function of synapses and, by extension, impairs learning and memory. Carrying this scenario over to humans, this process would happen either before neuritic plaques and neurofibrillary tangles are laid down, or simultaneously in a parallel process. In any event, it would occur outside our ability to measure it at this time. On separate occasions, both Mucke and LaFerla noted that their data call for treating, that is, inhibiting, both tau and Aβ accumulation as a therapeutic strategy.
The tau-transgenic mouse front had more news to offer in Washington. For example, the regulatable r4510Tg tau mouse studied by Karen Ashe’s and Brad Hyman’s labs is reported to develop tangles, neuronal loss, and a memory impairment that improves when soluble tau disappears even as tangles remain stable (SantaCruz et al., 2005). At the conference, these groups presented follow-up data suggesting a dissociation between those neurons that get tangles and those that die. In one study, the scientists used PHF1-staining of tau aggregates and cresyl violet staining of neurons for stereological counts in various subareas of hippocampus, striatum, and cortex of the r4510Tg tau mice to assess tau aggregation in relation to age and neuron loss. They describe varying proportions of hyperphosphorylated tau accumulation and cell death with age in these regions, but the upshot was that tau accumulation did not predict the pattern of neuron loss.
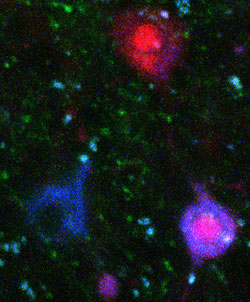
Tangles and Caspases
Multiphoton microscopy shows neurofibrillary tangles (blue) and caspases (red) in the brain of a live tau-transgenic mouse. [Photo courtesy of Tara Spires and Brad Hyman.]
Further, researchers in Hyman's lab have developed a fluorescent reporter that sticks to tangles and are using it with multiphoton microscopy to observe the natural history of tangle growth in live r4510Tg tau mice. First author Tara Spires also reported a way of monitoring caspase activation in vivo. Using RT-PCR, the authors noted an increase of caspase 8 expression in the brains of these mice. Of all the neurons the scientists found to have activated caspase 8, a majority did not have tangles. Taken together, these data hint at a caspase-dependent mechanism of cell death in connection with soluble tau in these mice.
If it’s not the tangles, what, then, is it about the biochemistry of tau that could disturb the function of neurons? Martin Ramsden in Ashe’s lab addresses this question by trying to isolate and characterize a 64 kDa, sarkosyl- insoluble form of tau the scientists hold responsible for the memory deficits in r4510Tg tau mice. Formal characterization of this strain was published this month (Ramsden et al., 2005).—Gabrielle Strobel.
See also Part 2, Part 3, and Part 4 of this series.
References
News Citations
- SfN: Return of the Other—Tau Is Back, Part 2
- SfN: Return of the Other—Tau Is Back, Part 3
- SfN: Return of the Other—Tau Is Back, Part 4
Paper Citations
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001 Jun 15;276(24):21562-70. PubMed.
- Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005 Jun 1;25(22):5446-54. PubMed.
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005 Jul 15;309(5733):476-81. PubMed.
- Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, Santacruz K, Guimaraes A, Yue M, Lewis J, Carlson G, Hutton M, Ashe KH. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci. 2005 Nov 16;25(46):10637-47. PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Spires TL, Orne JD, Santacruz K, Pitstick R, Carlson GA, Ashe KH, Hyman BT. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol. 2006 May;168(5):1598-607. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Banner Research Institute
A host of papers now indicates that neurofibrillary tangles (NFTs) are not a major cause of neuron death. These include an earlier paper by Brad Hyman as well as a paper by Renee Morsch, Bill Simon and me that concluded that neurons live for decades after they have formed NFTs. Although NFTs may not be a major cause of neuron death, they certainly contribute to decreased functional capacity of single neurons as well as synaptic deficits.
View all comments by Paul ColemanNew York State Institute for Basic Research
This is a well-described, interesting study. Together with studies mentioned above by P. Coleman, it is quite likely that neurofibrillary tangles (NFTs) of paired helical filaments (PHFs) themselves do not cause neuronal death in the diseased brain. Instead, formation of highly polymerized NFTs from the unpolymerized, abnormally hyperphosphorylated tau may be a defense mechanism of the affected neurons by which they turn the cytotoxic soluble hyperphosphorylated tau into inert NTFs (Alonso and Iqbal, 2005; Gong et al., 2006).
Several studies have demonstrated that unpolymerized hyperphosphorylated tau is neurotoxic, probably due to its sequestering normal tau and other microtubule-associated proteins and, thus, disrupting microtubules (Alonso et al., 1994; 1996; 1997; 2001; Fath et al., 2002), whereas upon polymerization into PHFs/NFTs, tau loses this toxic activity and becomes biologically and pathologically inert (Alonso et al., 2006). This may also explain why some NFT-containing neurons can survive for many years to decades (Morsch et al., 1999).
We have to distinguish the role between abnormally hyperphosphorylated tau and NFTs in the pathogenesis of tauopathies. This and other studies exclude a major role of NFTs in neuronal loss, but do not exclude such a role of the abnormally hyperphosphorylated tau. Actually, the hyperphosphorylated tau is toxic not only in vitro (Alonso et al., 1994; 1996; 1997; 2001), but also in vivo (Wittmann et al., 2001; Jackson et al., 2002). Hyperphosphorylation of tau at different phosphorylation sites has distinct effects on its loss of normal biological activity, its gain of toxic activity, and promoting tau’s self-assembly into PHFs. For example, phosphorylation sites at the middle part of tau (Ser199/Ser202/Thr205, Thr212, Thr231/Ser235, Ser262/Ser356) are critical to convert normal tau to a toxic molecule, whereas phosphorylation at the C-terminus of tau, including the PHF-1 sites (Ser396/Ser404), mainly promotes tau self-assembly into filaments (Abraha et al., 2000; Ferrari et al., 2003; Alonso et al., 2004; Haase et al., 2004). Because accumulated tau, both in human tauopathies and in all animal models of tauopathy, is always hyperphosphorylated, it is very possible that the neurodegeneration seen in these transgenic mouse brains results from unpolymerized hyperphosphorylated tau, whereas polymerization of this toxic tau into inert PHFs/NFTs is the neurons’ self-defense mechanism for survival.
Therefore, this study does not exclude a role of abnormal hyperphosphorylation of tau in neurodegeneration. Future studies on the correlation between time- and region-specific neurodegeneration and the level of unpolymerized hyperphosphorylation of tau, especially at those sites critical to toxicity, in this or transgenic mice with tauopathies, will no doubt provide new insight into the mechanism of tau-mediated neurodegeneration.
References:
Abraha A, Ghoshal N, Gamblin TC, Cryns V, Berry RW, Kuret J, Binder LI. C-terminal inhibition of tau assembly in vitro and in Alzheimer's disease. J Cell Sci. 2000 Nov;113 Pt 21:3737-45. PubMed.
del C Alonso A, Iqbal K. Tau-induced neurodegeneration: a clue to its mechanism. J Alzheimers Dis. 2005 Dec;8(3):223-6. PubMed.
Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A. 2001 Jun 5;98(12):6923-8. Epub 2001 May 29 PubMed.
Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996 Jul;2(7):783-7. PubMed.
Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A. 1994 Jun 7;91(12):5562-6. PubMed.
Alonso AD, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A. 1997 Jan 7;94(1):298-303. PubMed.
Alonso Ad, Li B, Grundke-Iqbal I, Iqbal K. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad Sci U S A. 2006 Jun 6;103(23):8864-9. PubMed.
Alonso Ad, Mederlyova A, Novak M, Grundke-Iqbal I, Iqbal K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem. 2004 Aug 13;279(33):34873-81. Epub 2004 Jun 9 PubMed.
Fath T, Eidenmüller J, Brandt R. Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer's disease. J Neurosci. 2002 Nov 15;22(22):9733-41. PubMed.
Ferrari A, Hoerndli F, Baechi T, Nitsch RM, Götz J. beta-Amyloid induces paired helical filament-like tau filaments in tissue culture. J Biol Chem. 2003 Oct 10;278(41):40162-8. PubMed.
Gong CX, Liu F, Grundke-Iqbal I, Iqbal K. Impaired brain glucose metabolism leads to Alzheimer neurofibrillary degeneration through a decrease in tau O-GlcNAcylation. J Alzheimers Dis. 2006 Mar;9(1):1-12. PubMed.
Haase C, Stieler JT, Arendt T, Holzer M. Pseudophosphorylation of tau protein alters its ability for self-aggregation. J Neurochem. 2004 Mar;88(6):1509-20. PubMed.
Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S, Geschwind DH. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 2002 May 16;34(4):509-19. PubMed.
Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol. 1999 Feb;58(2):188-97. PubMed.
Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001 Jul 27;293(5530):711-4. Epub 2001 Jun 14 PubMed.
View all comments by Cheng-Xin GongMake a Comment
To make a comment you must login or register.